Water–gas shift reaction
The water–gas shift reaction (WGSR) describes the reaction of carbon monoxide and water vapor to form carbon dioxide and hydrogen:
- CO + H2O ⇌ CO2 + H2
The water gas shift reaction was discovered by Italian physicist Felice Fontana in 1780. It was not until much later that the industrial value of this reaction was realized. Before the early 20th century, hydrogen was obtained by reacting steam under high pressure with iron to produce iron oxide and hydrogen. With the development of industrial processes that required hydrogen, such as the Haber–Bosch ammonia synthesis, a less expensive and more efficient method of hydrogen production was needed. As a resolution to this problem, the WGSR was combined with the gasification of coal to produce hydrogen. As the idea of hydrogen economy gains popularity, the focus on hydrogen as an energy storage medium when an alternative replacement energy source for hydrocarbons is used.
Applications
The WGSR is a highly valuable industrial reaction that is used in the manufacture of ammonia, hydrocarbons, methanol, and hydrogen. Its most important application is in conjunction with the conversion of carbon monoxide from steam reforming of methane or other hydrocarbons in the production of hydrogen.[1] In the Fischer–Tropsch process, the WGSR is one of the most important reactions used to balance the H2/CO ratio. It provides a source of hydrogen at the expense of carbon monoxide, which is important for the production of high purity hydrogen for use in ammonia synthesis.
The water–gas shift reaction may be an undesired side reaction in processes involving water and carbon monoxide, e.g. the rhodium-based Monsanto process. The iridium-based Cativa process uses less water, which suppresses this reaction.
Fuel cells
The WGSR can aid in the efficiency of fuel cells by increasing hydrogen production. The WGSR is considered a critical component in the reduction of carbon monoxide concentrations in cells that are susceptible to carbon monoxide poisoning such as the proton-exchange membrane (PEM) fuel cell.[2] The benefits of this application are two-fold: not only would the water gas shift reaction effectively reduce the concentration of carbon monoxide, but it would also increase the efficiency of the fuel cells by increasing hydrogen production.[2] Unfortunately, current commercial catalysts that are used in industrial water gas shift processes are not compatible with fuel cell applications.[3] With the high demand for clean fuel and the critical role of the water gas shift reaction in hydrogen fuel cells, the development of water gas shift catalysts for the application in fuel cell technology is an area of current research interest.
Catalysts for fuel cell application would need to operate at low temperatures. Since the WGSR is slow at lower temperatures where equilibrium favors hydrogen production, WGS reactors require large amounts of catalysts, which increases their cost and size beyond practical application.[2] The commercial LTS catalyst used in large scale industrial plants is also pyrophoric in its inactive state and therefore presents safety concerns for consumer applications.[3] Developing a catalyst that can overcome these limitations is relevant to implementation of a hydrogen economy.
Sorption enhanced water gas shift
The WGS reaction is used in combination with the solid adsorption of CO2 in the sorption enhanced water gas shift (SEWGS) in order to produce a high pressure hydrogen stream from syngas.[4]
Reaction conditions
The equilibrium of this reaction shows a significant temperature dependence and the equilibrium constant decreases with an increase in temperature, that is, higher hydrogen formation is observed at lower temperatures.
Temperature dependence
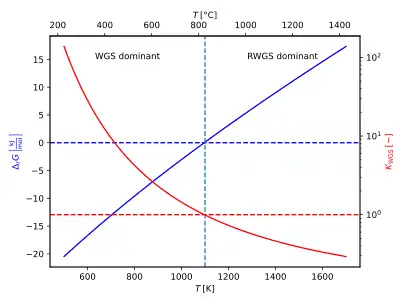
With increasing temperature, the reaction rate increases, but hydrogen production becomes less favorable thermodynamically[5] since the water gas shift reaction is moderately exothermic; this shift in chemical equilibrium can be explained according to Le Chatelier's principle. Over the temperature range of 600–2000 K, the equilibrium constant for the WGSR has the following relationship:[3]

Practical concerns
In order to take advantage of both the thermodynamics and kinetics of the reaction, the industrial scale water gas shift reaction is conducted in multiple adiabatic stages consisting of a high temperature shift (HTS) followed by a low temperature shift (LTS) with intersystem cooling.[6] The initial HTS takes advantage of the high reaction rates, but results in incomplete conversion of carbon monoxide. A subsequent low temperature shift reactor lowers the carbon monoxide content to <1%. Commercial HTS catalysts are based on iron oxide–chromium oxide and the LTS catalyst is a copper-based. The copper catalyst is susceptible to poisoning by sulfur. Sulfur compounds are removed prior to the LTS reactor by a guard bed. An important limitation for the HTS is the H2O/CO ratio where low ratios may lead to side reactions such as the formation of metallic iron, methanation, carbon deposition, and the Fischer–Tropsch reaction.
High temperature shift catalysis
The typical composition of commercial HTS catalyst has been reported as 74.2% Fe2O3, 10.0% Cr2O3, 0.2% MgO (remaining percentage attributed to volatile components).[7] The chromium acts to stabilize the iron oxide and prevents sintering. The operation of HTS catalysts occurs within the temperature range of 310 °C to 450 °C. The temperature increases along the length of the reactor due to the exothermic nature of the reaction. As such, the inlet temperature is maintained at 350 °C to prevent the exit temperature from exceeding 550 °C. Industrial reactors operate at a range from atmospheric pressure to 8375 kPa (82.7 atm).[7] The search for high performance HT WGS catalysts remains an intensive topic of research in fields of chemistry and materials science. Activation energy is a key criteria for the assessment of catalytic performance in WGS reactions. To date, some of the lowest activation energy values have been found for catalysts consisting of copper nanoparticles on ceria support materials,[8] with values as low as Ea = 34 kJ/mol reported relative to hydrogen generation.
Low temperature shift catalysis
Catalysts for the lower temperature WGS reaction are commonly based on copper or copper oxide loaded ceramic phases, While the most common supports include alumina or alumina with zinc oxide, other supports may include rare earth oxides, spinels or perovskites.[9] A typical composition of a commercial LTS catalyst has been reported as 32-33% CuO, 34-53% ZnO, 15-33% Al2O3.[3] The active catalytic species is CuO. The function of ZnO is to provide structural support as well as prevent the poisoning of copper by sulfur. The Al2O3 prevents dispersion and pellet shrinkage. The LTS shift reactor operates at a range of 200–250 °C. The upper temperature limit is due to the susceptibility of copper to thermal sintering. These lower temperatures also reduce the occurrence of side reactions that are observed in the case of the HTS. Noble metals such as platinum, supported on ceria, have also been used for LTS.[10]
Mechanism
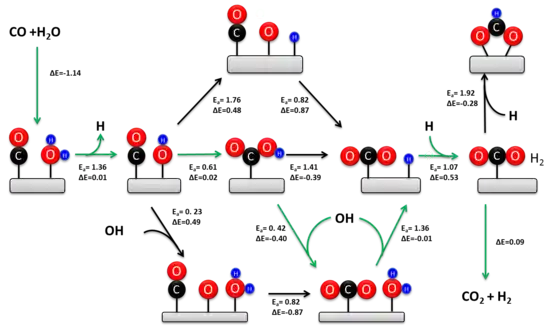
The WGSR has been extensively studied for over a hundred years. The kinetically relevant mechanism depends on the catalyst composition and the temperature.[6][14] Two mechanisms have been proposed: an associative Langmuir–Hinshelwood mechanism and a redox mechanism. The redox mechanism is generally regarded as kinetically relevant during the high-temperature WGSR (> 350 °C) over the industrial iron-chromia catalyst.[5] Historically, there has been much more controversy surrounding the mechanism at low temperatures. Recent experimental studies confirm that the associative carboxyl mechanism is the predominant low temperature pathway on metal-oxide-supported transition metal catalysts.[15][13]
Associative mechanism
In 1920 Armstrong and Hilditch first proposed the associative mechanism. In this mechanism CO and H2O are adsorbed onto the surface of the catalyst, followed by formation of an intermediate and the desorption of H2 and CO2. In general, H2O dissociates onto the catalyst to yield adsorbed OH and H. The dissociated water reacts with CO to form a carboxyl or formate intermediate. The intermediate subsequently dehydrogenates to yield CO2 and adsorbed H. Two adsorbed H atoms recombine to form H2.
There has been significant controversy surrounding the kinetically relevant intermediate during the associative mechanism. Experimental studies indicate that both intermediates contribute to the reaction rate over metal oxide supported transition metal catalysts.[15][13] However, the carboxyl pathway accounts for about 90% of the total rate owing to the thermodynamic stability of adsorbed formate on the oxide support. The active site for carboxyl formation consists of a metal atom adjacent to an adsorbed hydroxyl. This ensemble is readily formed at the metal-oxide interface and explains the much higher activity of oxide-supported transition metals relative to extended metal surfaces.[13] The turn-over-frequency for the WGSR is proportional to the equilibrium constant of hydroxyl formation, which rationalizes why reducible oxide supports (e.g. CeO2) are more active than irreducible supports (e.g. SiO2) and extended metal surfaces (e.g. Pt). In contrast to the active site for carboxyl formation, formate formation occurs on extended metal surfaces. The formate intermediate can be eliminated during the WGSR by using oxide-supported atomically dispersed transition metal catalysts, further confirming the kinetic dominance of the carboxyl pathway.[16]
Redox mechanism
The redox mechanism involves a change in the oxidation state of the catalytic material. In this mechanism, CO is oxidized by an O-atom intrinsically belonging to the catalytic material to form CO2. A water molecule undergoes dissociative adsorption at the newly formed O-vacancy to yield two hydroxyls. The hydroxyls disproportionate to yield H2 and return the catalytic surface back to its pre-reaction state.
Homogeneous models
The mechanism entails nucleophilic attack of water or hydroxide on a M-CO center, generating a metallacarboxylic acid.[2][17]
Thermodynamics
The WGSR is exergonic, with the following thermodynamic parameters at room temperature (298 K):
Free energy ΔG⊖ = –6.82 kcal Enthalpy ΔH⊖ = –9.84 kcal Entropy ΔS⊖ = –10.1 cal/deg
In aqueous solution, the reaction is less exergonic.[18]
Reverse water–gas shift
Since the water–gas shift reaction is an equilibrium reaction, there isn't a ‘reverse’ water–gas shift reaction. Water gas is defined as a fuel gas consisting mainly of carbon monoxide (CO) and hydrogen (H2). The term ‘shift’ in water–gas shift means changing the water gas composition (CO:H2) ratio. The ratio can be increased by adding CO2 or reduced by adding steam to the reactor.
See also
References
- Water Gas Shift Catalysis a combined experimental and computational study
- Vielstich, Wolf; Lamm, Arnold; Gasteiger, Hubert A., eds. (2003). Handbook of fuel cells: fundamentals, technology, applications. New York: Wiley. ISBN 978-0-471-49926-8.
- Callaghan, Caitlin (2006). Kinetics and catalysis of the water-gas-shift reaction: A Microkinetic and Graph Theoretic Approach (PDF) (PhD). Worcester Polytechnic Institute.
- Jansen, Daniel; van Selow, Edward; Cobden, Paul; Manzolini, Giampaolo; Macchi, Ennio; Gazzani, Matteo; Blom, Richard; Heriksen, Partow Pakdel; Beavis, Rich; Wright, Andrew (2013-01-01). "SEWGS Technology is Now Ready for Scale-up!". Energy Procedia. 37: 2265–2273. doi:10.1016/j.egypro.2013.06.107. ISSN 1876-6102.
- Ratnasamy, Chandra; Wagner, Jon P. (September 2009). "Water Gas Shift Catalysis". Catalysis Reviews. 51 (3): 325–440. doi:10.1080/01614940903048661. S2CID 98530242.
- Smith R J, Byron; Muruganandam Loganthan; Murthy Shekhar Shantha (2010). "A Review of the Water Gas Shift Reaction". International Journal of Chemical Reactor Engineering. 8: 1–32. doi:10.2202/1542-6580.2238. S2CID 96769998.
- Newsome, David S. (1980). "The Water-Gas Shift Reaction". Catalysis Reviews: Science and Engineering. 21 (2): 275–318. doi:10.1080/03602458008067535.
- Rodriguez, J.A.; Liu, P.; Wang, X.; Wen, W.; Hanson, J.; Hrbek, J.; Pérez, M.; Evans, J. (15 May 2009). "Water-gas shift activity of Cu surfaces and Cu nanoparticles supported on metal oxides". Catalysis Today. 143 (1–2): 45–50. doi:10.1016/j.cattod.2008.08.022.
- Coletta, Vitor C.; Gonçalves, Renato V.; Bernardi, Maria I. B.; Hanaor, Dorian A. H.; Assadi, M. Hussein N.; Marcos, Francielle C. F.; Nogueira, Francisco G. E.; Assaf, Elisabete M.; Mastelaro, Valmor R. (2021). "Cu-Modified SrTiO3 Perovskites Toward Enhanced Water–Gas Shift Catalysis: A Combined Experimental and Computational Study". ACS Applied Energy Materials. 4: 452–461. arXiv:2104.06739. doi:10.1021/acsaem.0c02371. S2CID 233231670.
- Jain, Rishabh; Maric, Radenka (April 2014). "Synthesis of nano-Pt onto ceria support as catalyst for water–gas shift reaction by Reactive Spray Deposition Technology". Applied Catalysis A: General. 475: 461–468. doi:10.1016/j.apcata.2014.01.053.
- Gokhale, Amit A.; Dumesic, James A.; Mavrikakis, Manos (2008-01-01). "On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper". Journal of the American Chemical Society. 130 (4): 1402–1414. doi:10.1021/ja0768237. ISSN 0002-7863. PMID 18181624.
- Grabow, Lars C.; Gokhale, Amit A.; Evans, Steven T.; Dumesic, James A.; Mavrikakis, Manos (2008-03-01). "Mechanism of the Water Gas Shift Reaction on Pt: First Principles, Experiments, and Microkinetic Modeling". The Journal of Physical Chemistry C. 112 (12): 4608–4617. doi:10.1021/jp7099702. ISSN 1932-7447.
- Nelson, Nicholas C.; Szanyi, János (2020-05-15). "Heterolytic Hydrogen Activation: Understanding Support Effects in Water–Gas Shift, Hydrodeoxygenation, and CO Oxidation Catalysis". ACS Catalysis. 10 (10): 5663–5671. doi:10.1021/acscatal.0c01059. OSTI 1656557. S2CID 218798723.
- Yao, Siyu; Zhang, Xiao; Zhou, Wu; Gao, Rui; Xu, Wenqian; Ye, Yifan; Lin, Lili; Wen, Xiaodong; Liu, Ping; Chen, Bingbing; Crumlin, Ethan (2017-06-22). "Atomic-layered Au clusters on α-MoC as catalysts for the low-temperature water-gas shift reaction". Science. 357 (6349): 389–393. Bibcode:2017Sci...357..389Y. doi:10.1126/science.aah4321. ISSN 0036-8075. PMID 28642235. S2CID 206651887.
- Nelson, Nicholas C.; Nguyen, Manh-Thuong; Glezakou, Vassiliki-Alexandra; Rousseau, Roger; Szanyi, János (October 2019). "Carboxyl intermediate formation via an in situ-generated metastable active site during water-gas shift catalysis". Nature Catalysis. 2 (10): 916–924. doi:10.1038/s41929-019-0343-2. ISSN 2520-1158. S2CID 202729116.
- Nelson, Nicholas C.; Chen, Linxiao; Meira, Debora; Kovarik, Libor; Szanyi, János (2020). "In Situ Dispersion of Palladium on TiO2 During Reverse Water–Gas Shift Reaction: Formation of Atomically Dispersed Palladium". Angewandte Chemie International Edition. 59 (40): 17657–17663. doi:10.1002/anie.202007576. ISSN 1521-3773. OSTI 1661896. PMID 32589820. S2CID 220118889.
- Barakat, Tarek; Rooke, Joanna C.; Genty, Eric; Cousin, Renaud; Siffert, Stéphane; Su, Bao-Lian (1 January 2013). "Gold catalysts in environmental remediation and water-gas shift technologies". Energy & Environmental Science. 6 (2): 371. doi:10.1039/c2ee22859a.
- King, A. D.; King, R. B.; Yang, D. B., "Homogeneous catalysis of the water gas shift reaction using iron pentacarbonyl", J. Am. Chem. Soc. 1980, vol. 102, pp. 1028-1032. doi:10.1021/ja00523a020