Hsp90 inhibitor
An Hsp90 inhibitor is a substance that inhibits that activity of the Hsp90 heat shock protein. Since Hsp90 stabilizes a variety of proteins required for survival of cancer cells, these substances may have therapeutic benefit in the treatment of various types of malignancies.[2] Furthermore, a number of Hsp90 inhibitors are currently undergoing clinical trials for a variety of cancers.[3] Hsp90 inhibitors include the natural products geldanamycin and radicicol as well as semisynthetic derivatives 17-N-Allylamino-17-demethoxygeldanamycin (17AAG).
| Hsp90 inhibitor | |
|---|---|
| Drug class | |
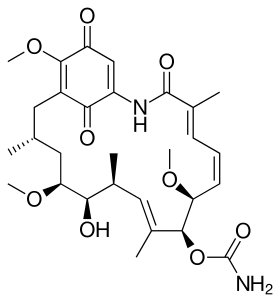 Geldanamycin, the first discovered Hsp90 inhibitor.[1] | |
| Class identifiers | |
| Use | Antineoplastic |
| Biological target | Hsp90 |
| Legal status | |
| In Wikidata | |
Mechanism of action
Among heat shock proteins the focus on HSP90 has increased due to its involvement in several cellular phenomena and more importantly in disease progression. HSP90 keeps the death proteins in an apoptosis resistant state by direct association. Its wide range of functions results from the ability of HSP90 to chaperone several client proteins that play a central pathogenic role in human diseases including cancer, neurodegenerative diseases and viral infection.[4] Geldanamycin directly binds to the ATP-binding pocket in the N-terminal domain of Hsp90 and, hence, blocks the binding of nucleotides to Hsp90. Analysis of the effects of Geldanamycin on steroid receptor activation indicates that the antibiotic blocks the chaperone cycle at the intermediate complex, preventing the release of the receptor from Hsp90 and, eventually, resulting in its degradation.[5] Ewing’s sarcoma shows several deregulated autocrine loops mediating cell survival and proliferation. So their blockade is a promising therapeutic approach. Proteosome analysis revealed that Hsp90 is differentially expressed between ewing’s sarcoma cell lines, sensitive and resistant to specific IGF1R/KIT inhibitors. The in vitro IGF1R/KIT pathway blockade on ewing’s sarcoma cell lines and classified ewing’s sarcoma cell lines as resistant and sensitive to blockade of pathway. Inhibition of Hsp90 with 17AAG and siRNA resulted in reduction of cell lines growth and survival. The inhibition of Hsp90 causes the proteosomal destruction of client proteins- Akt, KIT and IGF1R. This effect could be due to precluding physical contact between client proteins and Hsp90.[6] So since the molecular chaperones are overexpressed in a wide variety of cancer cells and in virally transformed cells, inhibiting the function of these chaperones is essential to controlling cancer cells, as this would affect the activity of signaling proteins. The availability of drugs that can specifically target Hsp90 and inhibit its function, resulting in the depletion of client proteins, has made Hsp90 a novel and exciting target for cancer therapy. HSP90 inhibitor NVP-BEP800 has been described to affect stability of SRC kinases clients and growth of T-cell and B-cell acute lymphoblastic leukemias. [7]
Natural product inhibitors
The first HSP90 inhibitors were developed from geldanamycin and radicicol which are the natural product inhibitors and are starting point for new approach.
HSP 90 is required for ATP dependent refolding of denatured or unfolded proteins and for the conformational maturation of a subset of proteins involved in the response of cells to extracellular signals. These include steroid receptors Raf – 1, Akt, Met and Her 2. HSP90 has conserved unique pocket in N terminal region. It binds ATP & ADP and has weak ATPase activity. This suggests that site acts as nucleotide or nucleotide ratio sensor. It is observed that nucleotides adopt unique C shaped bent shape when binding to this pocket. This is particularly unusual as nucleotides never adopt shape change in high affinity ATP/ADP sites. This also indicates that drugs that are developed should also have potential to adopt unique C shape conformation in order to bind the unique pocket. The rationale for this unusual need i.e. to bend the structure, is based on thermo dynamical fact that the molecule which needs minimum structural changes to go from unbound to bound state should not pay much entropic penalties and binding would be reflected by enthalpic factors.[8][9] Geldanamycin and radicicol tightly bind to this pocket and prevent the release of protein from chaperone complex. Thus the protein cannot achieve native conformation and is degraded by proteosome.[10] Addition of such inhibitor causes proteosomal degradation of signaling proteins like steroid receptors, Raf kinase and Akt. Geldanamycin and radicicol also inhibit mutated protein in cancer cells like P53, Vsrc, BCR – ABL. It is worth to note that the normal counterparts are not inhibited. Geldanamycin is an effective HSP90 inhibitor still it cannot be used in vivo because of its high toxicity and liver damage ability. The speculation is that the benzoquinone functional group is responsible. The semi-synthetic derivative 17 AAG, with lower toxicity but same potency as geldanamycin is developed and is currently under clinical trials.
Geldanamycin derivative 17 AAG
17-N-Allylamino-17-demethoxygeldanamycin (17AAG) is the semi-synthetic derivative of natural product Geldanamycin. It is less toxic with same therapeutic potential as Geldanamycin. It is the first HSP90 inhibitor to be evaluated in clinical trials. Currently 17AAG is being evaluated as potent drug against AML. It is known that 17 AAG decreases the concentration of client proteins but it was a question of debate if 17 AAG affected the genes for client proteins or it inhibited cytosolic proteins. Gene expression profiling of human colon cancer cell lines with 17AAG proves that Hsp90 client protein genes are not affected but the client proteins like hsc, keratin 8, keratin 18, akt, c-raf1 and caveolin-1 are deregulated resulting in inhibition of signal transduction.[11] Acute myelogenous leukemia (AML) remains the most common form of leukemia in the adult and elderly population. Currently, anthracyclines, cytarabine and etoposide are widely used in the treatment of AML due to their ability to induce apoptosis in leukemic cells. The signaling pathways by which these drugs work are not completely understood, but direct effects as DNA damage, mitochondrial electron transport interference, generation of oxidizing radicals and proteasomal activation have been demonstrated or hypothesized.[12] The 17-allylamino-17-demethoxygeldanamycin (17-AAG) derivative of GA is currently in clinical trial in cancer. Under normal conditions, Hsp90 acts on a wide range of client proteins and is essential for conformational maturation of numerous oncogenic signaling proteins, including protein kinases and ligand-regulated transcription factors. Hsp90 acts in a multiprotein complex with several co-chaperones. One of these, cochaperone p23, appears to stabilize Hsp90-complexes with steroid receptors and oncogenic tyrosine kinases. p23 also has chaperone activity on its own and is able to inhibit aggregation of denatured proteins in the absence of ATP. The ATP antagonist GA and its derivative 17AAG blocks p23 association with Hsp90, induces proteasomal degradation of survival signaling. Hsp90 client proteins, activates the apoptosis-associated double-stranded RNA-dependent protein kinase, PKR and promotes an apoptotic rather than a necrotic death type. p23 has increased expression in mammary carcinomas. In their study, Gausdal and colleagues found that anthracyclines and other chemotherapeutic drugs like cytarabine and etoposide, but not GA alone, induced caspase-dependent cleavage of p23. The cleavage could be catalyzed by either caspase-7 or caspase-3 and occurred at D142 or D145 in the C-terminal tail of p23 that is believed to be required for chaperone activity. The Hsp90 inhibitor GA was found to enhance caspase activation, p23 cleavage and apoptosis induced by anthracyclines. Finally they concluded that Hsp90, and consequently signaling mediated by client proteins in the Hsp90 multiprotein complex, may be targeted through p23 in chemotherapy-induced cell death in AML.[13][14]
Purine scaffolding
One of the important results obtained from the study of natural product inhibitor geldanamycin and its interaction with HSP90 is that the use of smaller molecules as inhibitors instead of complex molecules like radicicol is more efficient. Based on this information and advanced rational drug design technique, phenomenologically relevant scaffolds can be constructed. Random in vitro screening of library of small purine-related molecules led to identification and screening of more than 60000 compounds that have inhibition potency. Chiosis and colleagues reported the novel class of HSP90 inhibitors using rational design. The important factors considered in this rational design are
- Key interaction between inhibitor and Asp 93/ser 52 and lys 112/lys 58 at the base and top of the pocket respectively.
- Occupancy by inhibitor of hydrophobic pocket laying midway in the binding site and constituted by met98, val 150, leu 107, leu 103, phe 138 and val 186 is essential for affinity and selectivity.
- Molecules should have superior affinity to HSP90 as compared to natural nucleotides.
- Since many proteins depend on purine containing ligands for their function, derivatives of purine skeleton should have bioactivity, cell permeability and solubility.
So based on these considerations and observations Chiosis and colleagues theoretically designed following class of purines in which PU3 is the lead molecule. PU3 has a structural resemblance with ATP which is natural ligand for N terminal domain. X-ray crystallography data shows that PU3 has folded C shaped structure in both bound and free state. PU3 thus forms acceptable lead for further development of purine scaffold drugs. PU3 attaches to N terminal domain via the following key interactions.
- At the top 2 methoxy group of phenyl ring attaches itself to lys 112 of N terminal domain
- The 9 – N butyl chain occupy the lateral hydrophobic pocket. In fact this chain represents one of the most important elements of selectivity of PU3 for HSp90 versus similar pockets.
- At the base C6 amino group hydrogen bonds with asp93 – ser52[15]
Gamitrinib
Targeting networks of signaling pathways instead of single pathway is effective way for cancer treatment. Hsp90 is responsible for folding of proteins in multiple signaling networks in tumorigenesis. Mitochondrial Hsp90 is involved in complex signaling pathway that prevents initiation of induced apoptosis. Gamitrinib is a resorcinolic small molecule that specifically act on mitochondrial Hsp90. It induces a sudden loss of membrane potential which is followed by membrane rupture and initiation of apoptosis. Also gamitrinib is highly selective and does not affect normal cells.[9]
Future perspective
HSP90 is gaining increasing importance as a cancer target, in large part because of the potential for combinatorial targeting of multiple oncogenic protein pathways and biological effects. The good tolerability seen with the first-in-class drug 17-AAG has encouraged many biotechnology and large pharma companies to enter the field. The ability to demonstrate proof of concept for target modulation in patients has also been encouraging, as has the early evidence of clinical activity in melanoma 17-AAG is now in Phase II studies as a single agent and combination studies with cytotoxic and other agents such as the proteasome inhibitor bortezomib are also underway. Improved formulations for parenteral use are also being evaluated in the clinic. Radicicol-based inhibitors have not entered clinical development. Following on from the initial proof of concept studies with the natural product agents, considerable progress has been made in the preclinical development of small molecule, synthetic inhibitors, as exemplified by the purine and pyrazole based compounds. The recent rapid progress has built on a wealth of knowledge obtained with the natural product inhibitors and is a good example of the value of chemical biology studies in which the biological activity is identified first and then the molecular target is discovered by detailed biological studies. Current Medicinal Chemistry activities are focusing on the combined use of high throughput screening and structure-based design, coupled to the evaluation of the compounds in robust and mechanistically- informative biological assays. The next decade will be exciting in the HSP90 field as the clinical activity of the early geldanamycin-based drugs is rigorously evaluated while a series of synthetic small-molecule agents enter preclinical and clinical development. Particular areas of interest will include the potential for orally active HSP90 inhibitors and for the development of isoform-selective drugs that are targeted to particular members of the HSP90 family (DMAG –N-OXIDE). HSP90 inhibitors may also be evaluated in diseases other than cancer and where protein folding defects are involved in the disease pathology. It can be predicted that additional molecular chaperones will now be targeted for therapeutic intervention in cancer and other diseases. Furthermore, a portfolio of drugs can be envisaged that target various points in the protein quality control pathways of the malignant cell and other diseases states.
See also
References
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (August 1994). "Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation". Proc. Natl. Acad. Sci. U.S.A. 91 (18): 8324–8. doi:10.1073/pnas.91.18.8324. PMC 44598. PMID 8078881.
- Porter JR, Fritz CC, Depew KM (June 2010). "Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy". Curr Opin Chem Biol. 14 (3): 412–20. doi:10.1016/j.cbpa.2010.03.019. PMID 20409745.
- Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB (2009). "Update on Hsp90 inhibitors in clinical trial". Curr Top Med Chem. 9 (15): 1479–92. doi:10.2174/156802609789895728. PMC 7241864. PMID 19860730.
- Zhao R, Houry WA (December 2005). "Hsp90: a chaperone for protein folding and gene regulation". Biochem. Cell Biol. 83 (6): 703–10. doi:10.1139/o05-158. PMID 16333321.
- Hadden MK, Lubbers DJ, Blagg BS (2006). "Geldanamycin, radicicol, and chimeric inhibitors of the Hsp90 N-terminal ATP binding site". Curr Top Med Chem. 6 (11): 1173–82. doi:10.2174/156802606777812031. PMID 16842154.
- Martins AS, Ordoñez JL, García-Sánchez A, Herrero D, Sevillano V, Osuna D, Mackintosh C, Caballero G, Otero AP, Poremba C, Madoz-Gúrpide J, de Alava E (August 2008). "A pivotal role for heat shock protein 90 in Ewing sarcoma resistance to anti-insulin-like growth factor 1 receptor treatment: in vitro and in vivo study". Cancer Res. 68 (15): 6260–70. doi:10.1158/0008-5472.CAN-07-3074. PMID 18676850.
- Mshaik R, Simonet J, Georgievski A, Jamal L, Bechoua S, Ballerini P, Bellaye PS, Mlamla Z, Pais de Barros JP, Geissler A, Francin PJ, Girodon F, Garrido C, Quéré R (March 2021). "HSP90 inhibitor NVP-BEP800 affects stability of SRC kinases and growth of T-cell and B-cell acute lymphoblastic leukemias". Blood Cancer J. 3 (11): 61. doi:10.1038/s41408-021-00450-2. PMC 7973815. PMID 33737511.
- Chandrasekaran A, Pakkiriswami S, You JL, Acharya A, Packirisamy M, Maxwell D (2008). "Bioresistive identification of heat shock protein 90". Biomicrofluidics. 2 (3): 34102. doi:10.1063/1.2963104. PMC 2716925. PMID 19693369.
- Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC (March 2009). "Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90". J. Clin. Invest. 119 (3): 454–64. doi:10.1172/JCI37613. PMC 2648691. PMID 19229106.
- Lauria A, Ippolito M, Almerico AM (February 2009). "Inside the Hsp90 inhibitors binding mode through induced fit docking". J. Mol. Graph. Model. 27 (6): 712–22. doi:10.1016/j.jmgm.2008.11.004. PMID 19084447.
- Clarke PA, Hostein I, Banerji U, Stefano FD, Maloney A, Walton M, Judson I, Workman P (August 2000). "Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone". Oncogene. 19 (36): 4125–33. doi:10.1038/sj.onc.1203753. PMID 10962573.
- Al Shaer L, Walsby E, Gilkes A, Tonks A, Walsh V, Mills K, Burnett A, Rowntree C (May 2008). "Heat shock protein 90 inhibition is cytotoxic to primary AML cells expressing mutant FLT3 and results in altered downstream signalling". Br. J. Haematol. 141 (4): 483–93. doi:10.1111/j.1365-2141.2008.07053.x. PMID 18373709. S2CID 19960441.
- Gausdal G, Gjertsen BT, Fladmark KE, Demol H, Vandekerckhove J, Døskeland SO (December 2004). "Caspase-dependent, geldanamycin-enhanced cleavage of co-chaperone p23 in leukemic apoptosis". Leukemia. 18 (12): 1989–96. doi:10.1038/sj.leu.2403508. PMID 15483679.
- Piper PW, Millson SH, Mollapour M, Panaretou B, Siligardi G, Pearl LH, Prodromou C (December 2003). "Sensitivity to Hsp90-targeting drugs can arise with mutation to the Hsp90 chaperone, cochaperones and plasma membrane ATP binding cassette transporters of yeast". Eur. J. Biochem. 270 (23): 4689–95. doi:10.1046/j.1432-1033.2003.03866.x. PMID 14622256.
- Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N (October 2003). "Development of purine-scaffold small molecule inhibitors of Hsp90". Curr Cancer Drug Targets. 3 (5): 371–6. doi:10.2174/1568009033481778. PMID 14529388.