Isodicentric 15
Isodicentric 15, also called marker chromosome 15 syndrome,[2] idic(15), partial tetrasomy 15q, or inverted duplication 15 (inv dup 15), is a chromosome abnormality in which a child is born with extra genetic material from chromosome 15. People with idic(15) are typically born with 47 chromosomes in their body cells, instead of the normal 46. The extra chromosome, which is classified as a small supernumerary marker chromosome,[3] is made up of a piece of chromosome 15 that has been duplicated end-to-end like a mirror image. It is the presence of this extra genetic material that is thought to account for the symptoms seen in some people with idic(15). Individuals with idic(15) have a total of four copies of this chromosome 15 region instead of the usual two copies (1 copy each on the maternal and paternal chromosomes). The term isodicentric refers to a duplication and inversion of a centromere-containing chromosomal segment.[4]
| Isodicentric 15 | |
|---|---|
| Other names | Non-telomeric tetrasomy 15q[1] |
 | |
| An example of Isodicentric 15. The steps between 4x, 3x and 2x can be seen. | |
The syndrome is also often referred to by the term Chromosome 15q11.2-q13.1 Duplication Syndrome, shortened to Dup15q Syndrome, or marker chromosome 15 syndrome (mainly in the United States). Dup15q Syndrome includes both idic(15) and interstitial 15q11.2-q13.1, another type of duplication that causes similar clinical traits.
The extra chromosome is occasionally found in the mosaic state, i.e. some of the cells carry the marker chromosome. However, mostly because of the marker's instability and tendency to be lost during cell division (mitosis), some cells are completely normal with 46 chromosomes. Occasionally, cells may have more than one idic(15), resulting in 48 or 49 chromosomes in all or some of their cells. A similar clinical picture albeit to a milder degree could be expected in individuals that have the extra chromosome 15 material as an interstitial duplication (when the extra piece of chromosome 15 is included within the long arm of one of the two copies of chromosome 15, rather than as a small extra 'marker' chromosome) - often abbreviated to int dup(15); the individual thus having 46 chromosomes.[5][6]
Signs and symptoms
The severity of symptoms of idic(15) vary greatly between individuals. Individuals with idic(15) usually have delays in language development and motor skills such as walking or sitting up. Other traits may include low muscle tone (hypotonia), seizures (>50%), short stature, and intellectual disability. Distinctive facial features associated with idic(15) - where present at all - are usually very subtle but may include epicanthal folds (skin folds at the inner corners of one or both eyes), downward slanting palpebral fissures, broad forehead, a flattened nasal bridge, button nose, and a high arched palate (roof of the mouth). Some individuals show other signs that can often be associated with chromosomal conditions, such as pectus excavatum, or a unilateral or bilateral single transverse palmar crease. Many individuals with idic(15) display features of autism, such as problems with communication and social interactions, obsessional interests (often with interactive mechanisms like wheels, doors or switches), unpredictable sleep cycles (and a reduced need for sleep), and repetitive and stereotyped behaviors (e.g., lining up toys, playing with a toy in the same manner over and over again, hand flapping, rocking back and forth). Sensory processing is often affected, especially the vestibular system. A high pain threshold is often observed.[7] If speech develops, it is often echolalic but some individuals do grasp some language. With a severely affected person there may be an inability to walk or talk.
Genetics
Generally, idic(15) is not inherited; it is said to appear de novo, in one member of the family, by chance. In most cases, the abnormal chromosome is generated in the mother's germ cells: the oocytes. This finding is due to ascertainment bias; cases with maternally derived idic(15) usually have clinical findings and attract attention, but those with paternally derived idic(15) usually do not. Thus, diagnosed cases are usually patients where the duplicated material is derived from the mother's egg cell rather than the father's sperm cell.[8]
People with idic(15) have extra genetic material that has developed from chromosome 15. The material usually exists as a little extra chromosome 15; sometimes called a marker chromosome or an extra structurally abnormal chromosome (ESAC). The marker usually exists as an isodicentric chromosome; i.e. 2 copies of a specific part of the long arm of chromosome 15q11.2-q13.1 that is mirrored and doubled, with 2 centromeres and 2 DNA satellites. The smallest markers appear to be harmless and they may go undetected. However, if they are large enough to contain a number of important genes, they may result in "idic(15) syndrome" which is characterized by learning disabilities, autism and other neurological symptoms.[9] One of the regions responsible for the symptoms of idic(15) syndrome is the critical PWS/AS-region named after the Prader-Willi and/or Angelman syndromes.
Isodicentric chromosome 15 and autism
For more than 12 years, scientists have noticed that some individuals with autism also have idic(15). In fact, idic(15) is the most frequently identified chromosome problem in individuals with autism. (A chromosome anomaly involves extra or missing chromosomal material, not changes within the genes such as Fragile X syndrome). It is suggested that the co-occurrence of autism and idic(15) is not by chance. There may be a gene or genes in the 15q11-q13 region that is/are related to the development of autism in some individuals.
Genetic research studies of individuals without chromosome anomalies also support this idea that an autism-related gene may be present in 15q11.2-q13.1 Specifically, research studies found that certain DNA markers from the (15q1.2-q13.1) region were found more often in individuals with autism than in individuals without autism. Although these DNA markers are too small to be genes, they suggest that researchers may be getting close to finding an autism gene in this region.[10]
A recent study reported the introduction of two extra copies of just a single gene present in the 15q11.2-q13.1 region, Ube3a, into mice to model the gene copy number expressed in the brain in idic(15).[11] These mice displayed autism-related behavioral deficits including impaired social interaction, reduced ultrasonic vocal communication, and increased repetitive behavior (self-grooming).[12]
Diagnosis
The extra chromosome in people with idic(15) can be easily detected through chromosome analysis (karyotyping). Additional tests are usually required. FISH (Fluorescent in situ hybridization) is used to confirm the diagnosis by distinguishing idic(15) from other supernumerary marker chromosomes. Array CGH can be used to determine the gene content and magnitude of copy number variation so that the clinical picture can be foreseen.[13] Interstitial duplications of chromosome 15 can be more difficult to detect on a routine chromosome analysis but are clearly identifiable using a 15q FISH study. Families should always discuss the results of chromosome and FISH studies with a genetic counselor or other genetics professionals to ensure accurate interpretation.
Screening
In general, idic(15) occurs de novo but the parents must be karyotyped to make sure it is not inherited, mostly because this will affect the course of genetic counseling given to the family. If the abnormality is found prenatally and one of the parents harbour the marker, the child has a chance of not carrying the mutation. Further tests should however be done to prove the marker has not been rearranged while being inherited. This information is also necessary for counseling of future pregnancies. Each family is unique and should therefore be handled individually.[9]
Management
At the present time, there is no specific treatment that can undo any chromosomal abnormality, nor the genetic pattern seen in people with idic(15). The extra chromosomal material in those affected was present at or shortly after conception, and its effects on brain development began taking place long before the child was born. Therapies are available to help address many of the symptoms associated with idic(15). Physical, occupational, and speech therapies along with special education techniques can stimulate children with idic(15) to develop to their full potential.
In terms of medical management of the symptoms associated with Chromosome 15q11.2-q13.1 Duplication Syndrome, families should be aware that individuals with chromosome 15 duplications may tolerate medications differently and may be more sensitive to side effects for some classes of medications, such as the serotonin reuptake inhibitor type medications (SSRI).[14] Thus, these should be used with caution and any new medication should be instituted in a controlled setting, with slow titration of levels and with a clear endpoint as to what the expected outcome for treatment is.
There is an increased risk of sudden, unexpected death among children and adults with this syndrome. The full cause is not yet understood but it is generally attributed to SUDEP (Sudden Unexplained Death in Epilepsy).[15]
Epidemiology
About half of all 'marker' chromosomes are idic(15) but idic(15) in itself is one of the rare chromosome abnormalities. Incidence at birth appears to be 1 in 30,000[16] with a sex ratio of almost 1:1; however, since dysmorphic features are absent or subtle and major malformations are rare, chromosome analysis may not be thought to be indicated, and some individuals, particularly in the older age groups, probably remain undiagnosed.[16] There are organizations for families with idic(15) children that offer extensive information and support.[6]
Research
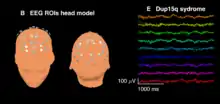
Patients with idic(15) and int dup(15) often feature a distinctive electroencephalography (EEG) signature or biomarker in the form of high amplitude spontaneous beta frequency (12–30 Hz) oscillations. This EEG signature was first noted as a qualitative pattern in clinical EEG readings and was later described quantitatively by researchers at the University of California, Los Angeles and their collaborators within the network of national Dup15q clinics.[17] This group of researchers found that beta activity in children with Dup15q syndrome is significantly greater than that observed in (1) healthy, typically developing children of the same age and (2) children of the same age and IQ with autism not caused by a known genetic disorder (i.e., nonsyndromic ASD). The EEG signature appears almost identical to beta oscillations induced by benzodiazepine drugs that modulate GABAA receptors, suggesting that the signature is driven by overexpression of duplicated GABAA receptor genes GABRA5, GABRB3, and GABRG3 found on 15q11.2-q13.1. Treatment monitoring and identification of molecular disease mechanisms may be facilitated by this biomarker.
See also
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Duplication/inversion 15q11". www.orpha.net. Retrieved 22 May 2019.
- Jafari-Ghahfarokhi H, Moradi-Chaleshtori M, Liehr T, Hashemzadeh-Chaleshtori M, Teimori H, Ghasemi-Dehkordi P (2015). "Small supernumerary marker chromosomes and their correlation with specific syndromes". Advanced Biomedical Research. 4: 140. doi:10.4103/2277-9175.161542. PMC 4544121. PMID 26322288.
- Chen CP, Lin HY, Wang LK, Chern SR, Wu PS, Chen SW, Wu FT, Fran S, Chen YY, Town DD, Pan CW, Wang W (July 2020). "Prenatal diagnosis and molecular cytogenetic characterization of a small supernumerary marker chromosome derived from inv dup(15)". Taiwanese Journal of Obstetrics & Gynecology. 59 (4): 580–585. doi:10.1016/j.tjog.2020.05.019. PMID 32653133.
- Gardner, R. J. M. (2012). Chromosome abnormalities and genetic counseling. Grant R. Sutherland, Lisa G. Shaffer (4th ed.). Oxford: Oxford University Press. p. 306. ISBN 978-0-19-974915-7. OCLC 769344040.
- Hogart, Amber; Wu, David; Lasalle, Janine M.; Schanen, N. Carolyn (2008). "The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13.1". Neurobiology of Disease. 38 (2): 181–191. doi:10.1016/j.nbd.2008.08.011. PMC 2884398. PMID 18840528.
- "IDEAS - Isodicentric 15 | Welcome to IDEAS: IsoDicentric 15 Exchange Advocacy and Support | Support and Advocacy for duplication 15q syndrome: IDIC15, IDIC(15), Interstitial d..." Archived from the original on June 4, 2008.
- Luchsinger, Kadi; Lau, Heather; Hedlund, Julie L; Friedman, Daniel; Krushel, Kara; Devinsky, Orrin (2016). "Parental-reported pain insensitivity in Dup15q". Epilepsy & Behavior. 55: 124–7. doi:10.1016/j.yebeh.2015.10.007. PMID 26773682. S2CID 32626204.
- Cook E.H. Jr; Lindgren V.; Leventhal B.L.; Courchesne R.; Lincoln A.; Shulman C.; Lord C.; Courchesne E. (1997). "Autism or atypical autism in maternally but not paternally derived proximal 15q duplication". Am. J. Hum. Genet. 60 (4): 928–934. PMC 1712464. PMID 9106540.
- RJ McKinlay Gardner, Grant R. Sutherland. Chromosome Abnormalities and Genetic Counseling, 3rd Ed, Oxford University Press, New York 2004. ISBN 0-19-514960-2
- http://www.exploringautism.org/autism/iso_chr15.htm
- Smith, S. E. P.; Zhou, Y.-D.; Zhang, G.; Jin, Z.; Stoppel, D. C.; Anderson, M. P. (2011). "Increased Gene Dosage of Ube3a Results in Autism Traits and Decreased Glutamate Synaptic Transmission in Mice". Science Translational Medicine. 3 (103): 103ra97. doi:10.1126/scitranslmed.3002627. PMC 3356696. PMID 21974935.
- "Science Translational Medicine: Making Mice with Autistic Behavior".
- Wang N.J.; Liu D.; Parokonny A.S.; Schanen N.C. (2004). "High-resolution molecular characterization of 15q11–q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage". Am. J. Hum. Genet. 75 (2): 267–281. doi:10.1086/422854. PMC 1216061. PMID 15197683.
- Schanen, C: Research update on chromosome 15 duplications – idic(15) and interstitial duplications: The duplication 15q syndrome. Presentation at 2005 International Conference on Isodicentric 15 and Related Disorders.
- "Physician Advisory - Dup15q". 8 April 2014.
- Battaglia A (2008). "The inv dup (15) or idic (15) syndrome (Tetrasomy 15q)". Orphanet J Rare Dis. 3: 30. doi:10.1186/1750-1172-3-30. PMC 2613132. PMID 19019226.
- Frohlich J., Senturk D., Saravanapandian V., Golshani P., Reiter L.T., Sankar R., Thibert R.L., DiStefano C., Huberty S., Cook E.H., Jeste S.S. (2016). "A Quantitative Electrophysiological Biomarker of Duplication 15q11. 2-q13. 1 Syndrome". PLOS ONE. 11 (12): e0167179. Bibcode:2016PLoSO..1167179F. doi:10.1371/journal.pone.0167179. PMC 5157977. PMID 27977700.
{{cite journal}}: CS1 maint: multiple names: authors list (link)