IPEX syndrome
Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX syndrome) is a rare autoimmune disease. It is one of the autoimmune polyendocrine syndromes. Most often, IPEX presents with autoimmune enteropathy, dermatitis (eczema), and autoimmune endocrinopathy (most often Type 1 diabetes), but other presentations exist.[5]
| IPEX syndrome | |
|---|---|
| Other names | Autoimmune enteropathy type 1[1] |
 | |
| IPEX syndrome is inherited via X-linked recessive | |
| Specialty | Immunology |
| Symptoms | Lymphadenopathy[2] |
| Causes | FOXP3 gene mutation[1] |
| Diagnostic method | Family history, Genetic test[1] |
| Treatment | TPN(nutritional purpose), Cyclosporin A and FK506, Bone marrow transplant[3][4] |
IPEX is caused by mutations in the gene FOXP3, which encodes transcription factor forkhead box P3 (FOXP3). FOXP3 is widely considered to be the master regulator of the regulatory T cell (Treg) lineage.[6][7] FOXP3 mutation can lead to the dysfunction of CD4+ Tregs. In healthy people, Tregs maintain immune homeostasis.[8] When there is a deleterious FOXP3 mutation, Tregs do not function properly and cause autoimmunity.[8][9]
IPEX onset usually happens in infancy. If left untreated, it is often fatal by the age of 2 or 3.[10][11] A bone marrow transplant is generally considered the best treatment option.[11] IPEX exclusively affects males and is inherited in an X-linked recessive manner;[1][2] female carriers of pathogenic FOXP3 mutations do not have symptoms and no female cases are known.[4]
Presentation
Classical triad
The classical triad describes the most common symptoms of IPEX: intractable diarrhea, type 1 diabetes, and eczema. Symptoms usually begin shortly after birth.[12]
Other symptoms include: thyroid disease, kidney dysfunction, blood disorders, frequent infections, autoimmune hemolytic anemia, and food allergies, among others.[10]
Endocrinopathy
The most common endocrinopathy associated with IPEX is type 1 diabetes, especially neonatal diabetes. In this type of diabetes, the immune system attacks insulin-producing cells. This makes the pancreas unable to produce insulin. Diabetes can permanently damage the pancreas.[13]
Thyroid disorders are also common.[14]
Enteropathy
The most common enteropathy associated with IPEX is intractable diarrhea. Vomiting and gastritis are also common. Other manifestations include Celiac disease, ulcerative colitis, and ileus.[14]

Skin manifestations
The most common form of skin involvement is dermatitis. It can occur in three forms: eczematiform (mainly atopic dermatitis), ichthyosiform, psoriasiform, or a combination. Other skin manifestations can include cheilitis, onychodystrophy, and alopecia.[14]
Early life
IPEX patients are usually born with normal weight and length at term. Nevertheless, the first symptoms may present in the first days of life,[15] and some reported cases labeled newborns with intrauterine growth restriction and evidence of meconium in the amniotic fluid.[16]
Genetics
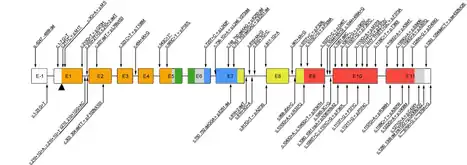
FOXP3 gene
IPEX syndrome is inherited in males in an X-linked recessive pattern through the FOXP3 gene. FOXP3's cytogenetic location is Xp11.23.[6][7] The FOXP3 gene has 12 exons and its full reading open frame encodes 431 amino acids. FOXP3 is a member of the FKH family of transcription factors and contains a proline‐rich (PRR) amino‐terminal domain, central zinc finger (ZF) and leucine zipper (LZ) domains important for protein–protein interactions. It also has a carboxyl‐terminal FKH domain required for nuclear localization and DNA‐binding activity. In humans, exons 2 and 7 may be spliced and excluded from the protein.[17]
FOXP3 mutations
A large variety of mutations have been found, including single base substitutions, deletions, and splicing mutations. Data from 2018 describes over 70 mutations in the FOXP3 gene leading to IPEX syndrome. This number has grown dramatically in the past decade.[18] In 2010 there were only 20 mutations of FOXP3 known in the literature.[19] Some mutations cause FOXP3 expression to malfunction, which leads to a defect in Treg production. Those individuals do not have circulating CD4+/CD25+/FOXP3+ Treg cells. Reduced expression of FOXP3 has been described, and these individuals may express normal levels of dysfunctional protein, which leads to mild symptoms during the neonatal period or later in life. Other individuals express no FOXP3 protein.[20] A common location for mutation of FOXP3 leading to expression of malfunctioning protein is the DNA-binding domain called the forkhead domain. The mutation makes the truncated protein unable to bind to its DNA binding site. This impairs its function concerning Treg development and functioning. The absence or dysfunction of Tregs causes autoimmune symptoms.[19]
FOXP3 pathways
FOXP3 can function as both a repressor and a trans‐activator of Treg cells depending on its interactions with other proteins. FOXP3 expression is characterised by controlling transcription, influencing epigenetic changes and post-transcriptional modifications. The N‐terminal repressor domain of FOXP3 can change transcription or epigenetic regulation of Treg cells. Transcriptional activity is altered through interactions between the N-terminal domain and Eos - which associates with CtBP1 and forms a corepressor complex. This complex binds the IL2 promoter and enables FOXP3 to repress IL2 transcription in Treg cells. FOXP3 forms complexes with histone deacetylase (HDAC)7, HDAC9, and the histone acetyl transferase TIP60, which alters epigenetic activity of Treg cells. The N‐terminal domain of FOXP3 can also antagonize the transcription factors RORγ and RORα, thereby inhibiting TH17 cell differentiation. FOXP3 is linked to TCR signaling by downstream transcription factors. All of these findings verify the importance of FOXP3 in the regulation of transcriptional activity and repression in Treg cells.[17]
Diagnosis
Early detection of the disease is crucial because IPEX has a high mortality level if left untreated.[20] IPEX is usually diagnosed based on the following criteria:[1][4]
- Clinical triad
- Family history
- Laboratory findings: elevated serum concentration of IgE, eosinophilia, autoimmune anemia and decreased number of FOXP3 Treg cells.
- Genetic testing: single-gene testing and multigene panel.
Treatment
Individuals with IPEX will usually need supportive care in a hospital. Most common is nutritional treatment for enteropathy and insulin therapy for T1D. IPEX treatment tends to focus on managing symptoms, reducing autoimmunity, and/or treating secondary conditions. Usually, treatment will involve immunosuppression.[11]
Drugs used include:
Currently, the standard treatment for IPEX is a bone marrow transplant. If donor-recipient chimerism is achieved, individuals with IPEX can achieve complete remission.[11]
Research
In 1982, Powel et al. published a case report of a family with 19 males who were affected by an X-linked syndrome with symptoms including polyendocrinopathy and diarrhea. The most common symptoms in this family were severe enteropathy, T1D, and dermatitis. Only 2 of the 19 affected males in the family survived past 3 years old. These individuals lived to 10 and 30 years old.[21] Powel's study is now widely considered the first documentation of IPEX.
Scurfy mouse
Scurfy is a type of model mouse used for immunology research. Scurfy mice have had 2 base pairs inserted within the FOXP3 gene. This leads to a frameshift mutation in FOXP3 gene and the expressed protein is truncated, causing functional deficiency of Treg cells. Then, autoreactive CD4+T cells and inflammatory cells cause tissue damage.[22] Scurfy mice have an enlarged spleen and lymph nodes, squinted red eyes, and scaly or "ruffled" skin. The mice also have immunity problems and tend to die approximately 3 weeks after birth.[18] From 2000 - 2001, multiple studies confirmed that IPEX is the human equivalent of scurfy mice and that the FOXP3 gene is responsible.[10]
References
- "Orphanet: Immune dysregulation polyendocrinopathy enteropathy X linked syndrome". www.orpha.net. Retrieved 2017-04-18.
- "Immunodysregulation, polyendocrinopathy and enteropathy X-linked | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2019-01-09. Retrieved 2017-04-16.
- Eisenbarth GS (2010-12-13). Immunoendocrinology: Scientific and Clinical Aspects. Springer Science & Business Media. pp. 129–138. ISBN 9781603274784.
- Hannibal M, Torgerson T (1993-01-01). "IPEX Syndrome". In RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Ledbetter N, Mefford H (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301297.update 2011
- "Embase". www.embase.com. Retrieved 2023-04-04.
- "IPEX syndrome". Genetics Home Reference. Retrieved 2017-04-16.
- "FOXP3 gene". Genetics Home Reference. Retrieved 2017-04-16.
- Huang, Qianru; Liu, Xu; Zhang, Yujia; Huang, Jingyao; Li, Dan; Li, Bin (2020-01-20). "Molecular feature and therapeutic perspectives of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome". Journal of Genetics and Genomics. 47 (1): 17–26. doi:10.1016/j.jgg.2019.11.011. ISSN 1673-8527. PMID 32081609. S2CID 211231953.
- Bacchetta, Rosa; Barzaghi, Federica; Roncarolo, Maria-Grazia (April 2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation: IPEX syndrome and FOXP3". Annals of the New York Academy of Sciences. 1417 (1): 5–22. Bibcode:2018NYASA1417....5B. doi:10.1111/nyas.13011. PMID 26918796. S2CID 11149833.
- Bacchetta, Rosa; Barzaghi, Federica; Roncarolo, Maria-Grazia (2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation: IPEX syndrome and FOXP3". Annals of the New York Academy of Sciences. 1417 (1): 5–22. Bibcode:2018NYASA1417....5B. doi:10.1111/nyas.13011. PMID 26918796. S2CID 11149833.
- Ben-Skowronek, Iwona (2021). "IPEX Syndrome: Genetics and Treatment Options". Genes. 12 (3): 323. doi:10.3390/genes12030323. ISSN 2073-4425. PMC 7995986. PMID 33668198.
- Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. (June 2020). "Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review". Autoimmunity Reviews. 19 (6): 102526. doi:10.1016/j.autrev.2020.102526. PMID 32234571. S2CID 214768010.
- CDC (2022-03-11). "What Is Type 1 Diabetes?". Centers for Disease Control and Prevention. Retrieved 2023-04-16.
- Park, Jae Hyon; Lee, Keum Hwa; Jeon, Bokyoung; Ochs, Hans D.; Lee, Joon Suk; Gee, Heon Yung; Seo, Seeun; Geum, Dongil; Piccirillo, Ciriaco A.; Eisenhut, Michael; van der Vliet, Hans J.; Lee, Jiwon M.; Kronbichler, Andreas; Ko, Younhee; Shin, Jae Il (2020-06-01). "Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review". Autoimmunity Reviews. 19 (6): 102526. doi:10.1016/j.autrev.2020.102526. ISSN 1568-9972. PMID 32234571. S2CID 214768010.
- Barzaghi F, Passerini L, Bacchetta R (2012). "Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity". Frontiers in Immunology. 3: 211. doi:10.3389/fimmu.2012.00211. PMC 3459184. PMID 23060872.
- Xavier-da-Silva MM, Moreira-Filho CA, Suzuki E, Patricio F, Coutinho A, Carneiro-Sampaio M (February 2015). "Fetal-onset IPEX: report of two families and review of literature". Clinical Immunology. 156 (2): 131–40. doi:10.1016/j.clim.2014.12.007. PMID 25546394.
- Bacchetta, Rosa; Barzaghi, Federica; Roncarolo, Maria-Grazia (2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation". Annals of the New York Academy of Sciences. 1417 (1): 5–22. Bibcode:2018NYASA1417....5B. doi:10.1111/nyas.13011. ISSN 1749-6632. PMID 26918796.
- Bacchetta R, Barzaghi F, Roncarolo MG (April 2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation". Annals of the New York Academy of Sciences. 1417 (1): 5–22. Bibcode:2018NYASA1417....5B. doi:10.1111/nyas.13011. PMID 26918796.
- Michels AW, Gottlieb PA (May 2010). "Autoimmune polyglandular syndromes". Nature Reviews. Endocrinology. 6 (5): 270–7. doi:10.1038/nrendo.2010.40. PMID 20309000. S2CID 20395564.
- Kitcharoensakkul, Maleewan; Cooper, Megan A. (2020-01-01), Rose, Noel R.; Mackay, Ian R. (eds.), "Chapter 28 - Autoimmunity in Primary Immunodeficiency Disorders", The Autoimmune Diseases (Sixth Edition), Academic Press, pp. 513–532, doi:10.1016/b978-0-12-812102-3.00028-2, ISBN 978-0-12-812102-3, S2CID 208377306, retrieved 2021-01-29
- Powell, Berkley R.; Buist, Neil R.M.; Stenzel, Peter (1982). "An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy". The Journal of Pediatrics. 100 (5): 731–737. doi:10.1016/s0022-3476(82)80573-8. ISSN 0022-3476. PMID 7040622.
- Yilmaz OK, Haeberle S, Zhang M, Fritzler MJ, Enk AH, Hadaschik EN (2019). "Scurfy Mice Develop Features of Connective Tissue Disease Overlap Syndrome and Mixed Connective Tissue Disease in the Absence of Regulatory T Cells". Frontiers in Immunology. 10: 881. doi:10.3389/fimmu.2019.00881. PMC 6491778. PMID 31068947.
Further reading
- Bacchetta R, Barzaghi F, Roncarolo MG (April 2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation". Annals of the New York Academy of Sciences. 1417 (1): 5–22. Bibcode:2018NYASA1417....5B. doi:10.1111/nyas.13011. PMID 26918796.
- Barzaghi F, Passerini L, Bacchetta R (1 January 2012). "Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity". Frontiers in Immunology. 3: 211. doi:10.3389/fimmu.2012.00211. PMC 3459184. PMID 23060872.
- Elzouki AY, Harfi HA, Nazer H, Stapleton FB, Oh W, Whitley RJ (2012-01-10). Textbook of Clinical Pediatrics. Springer Science & Business Media. ISBN 9783642022029.