Ionic hydrogenation
Ionic hydrogenation refers to hydrogenation achieved by the addition of a hydride to substrate that has been activated by an electrophile. Some ionic hydrogenations entail addition of H2 to the substrate and some entail replacement of a heteroatom with hydride.[1] Traditionally, the method was developed for acid-induced reductions with hydrosilanes. Alternatively ionic hydrogenation can be achieved using H2. [2] Ionic hydrogenation is employed when the substrate can produce a stable carbonium ion. Polar double bonds are favored substrates.
Using hydrosilanes
Because silicon (1.90) is more electropositive than hydrogen (2.20), hydrosilanes exhibit (mild) hydridic character. Hydrosilanes can serve as hydride donors to highly electrophilic organic substrates. Many alcohols, alkyl halides, acetals, orthoesters, alkenes, aldehydes, ketones, and carboxylic acid derivatives are suitable substrates. Such reactions often require Lewis acids. Only reactive electrophiles undergo reduction, selectivity is possible in reactions of substrates with multiple reducible functional groups.
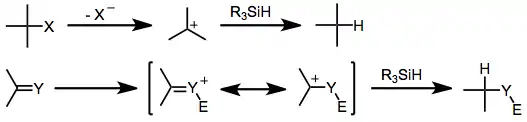
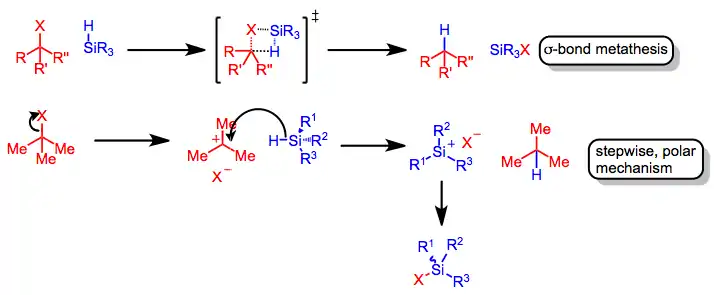
Upon the generation of a carbocation, rate-determining hydride transfer from the organosilane occurs to yield a reduced product. Retention of configuration at silicon has been observed in silane reductions of chiral triaryl methyl chlorides in benzene. This result suggests that the exchange of chlorine for hydrogen occurs through σ-bond metathesis.[3] Reductions in more polar solvents may involve silicenium ions.[4]
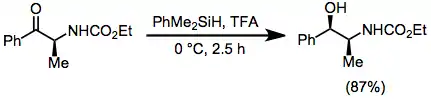
Polymeric hydrosilanes, such as polymethylhydrosiloxane (PHMS) may be employed to facilitate separation of the reduced products from silicon-containing byproducts.[5][6]
Using H2
The proton and hydride transfers are usually sequential or concerted. Usually ionic hydrogenation is shown to occur in two steps, starting with protonation.
- R2C=Y + H+ → R2C+-YH
- R2C+-YH + "H−" → R2CH-YH
Substrates
In the case of metal-catalyzed ionic hydrogenation, the substrates and their products must not bind to metal sites, as this would interfere with H2 activation. Ketones are the most common substrates.[7] Less common are imines and N-heterocycles. The reaction can also be performed in reverse to effect hydrogenolysis. Liquid substrates can sometimes be hydrogenated without solvent, a goal of green chemistry.[2]
Proton and hydride pairs
The most common hydrogenating pair is an organosilane as the hydride source (e.g. triethylsilane), and a strong oxyacid as the proton source (e.g. trifluoroacetic acid or triflic acid). The hydride and proton source cannot combine to give H2, which limits the hydricity and acidity of the H− and H+ sources, respectively.
Transition metal hydride complexes can be used in place of organosilanes as the hydride source. In these cases, triflic acid is a typical proton donor. Ketones such as benzophenones, and 1,1-disubstituted olefins are typical substrates. Hydrides of tungsten, chromium, osmium, and molybdenum complexes have also been reported. Tungsten dihydride complexes can hydrogenate ketones stoichiometrically with no external acids. One hydride serves as the hydride source, and the other serves as a proton source.[2]
In the case of ionic hydrogenation, a dihydride complex is regenerated by hydrogen gas following hydrogenation. Typical catalysts are tungsten or molybdenum complexes. An example of such a catalyst is CpMo(CO)2(PR3)(OCR'2)]+ where M = W or Mo.[7]
Related reactions
Transfer hydrogenation (TH) catalysts, e.g. Shvo catalyst, are related to catalysts used for ionic hydrogenation. TH catalysts however do not employ strong acids and both the H− and H+ components are covalently bonded to the complex prior to transfer to the unsaturated substrates. Typically, TH catalysts are more widely employed in organic synthesis.[8]
Older literature
- Kursanov, D. N.; Parnes, Z. N.; Loim, N. M. (1974). "Applications of Ionic Hydrogenation to Organic Synthesis". Synthesis. 1974 (9): 633–651. doi:10.1055/s-1974-23387.
References
- Colvin, Ernest W. (1981). "Silanes as Reducing Agents". Silicon in Organic Synthesis. pp. 325–336. doi:10.1016/B978-0-408-10831-7.50027-5. ISBN 9780408108317.
- Bullock, R. M. "Ionic Hydrogenations," in The Handbook of Homogeneous Hydrogenation (eds J. G. de Vries and C. J. Elsevier), Wiley-VCH Verlag GmbH, Weinheim, Germany, 2007. doi:10.1002/9783527619382.ch7
- Austin, J. D.; Eaborn, C. J. Chem. Soc. 1964, 2279.
- Sommer, L. H.; Bauman, D. L. J. Am. Chem. Soc. 1969, 91, 7045.
- Pri-Bar, I.; Buchman, O. J. Org. Chem. 1986, 51, 734.
- Fujita, M.; Hiyama, T. J. Org. Chem. 1988, 53, 5415.
- Bullock, R. Morris (17 May 2004). "Catalytic Ionic Hydrogenations". Chemistry: A European Journal. 10 (10): 2366–2374. doi:10.1002/chem.200305639. PMID 15146510.
- Eisenstein, O.; Crabtree, R. H., "Outer sphere hydrogenation catalysis", New J. Chem. 2013, vol. 37, 21 doi:10.1039/c2nj40659d