L1 syndrome
L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS).[1][2] It is also called L1CAM syndrome (for the disorder's causative gene) and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation (intellectual disability), adducted thumbs, spasticity, and hydrocephalus.[2]
| L1 syndrome | |
|---|---|
| Other names | L1CAM syndrome, CRASH syndrome, Corpus callosum hypoplasia-retardation-adducted thumbs-spasticity-hydrocephalus syndrome |
 | |
| Specialty | Pediatrics, neurology, medical genetics |
| Usual onset | Neonatal |
| Duration | Lifelong |
| Risk factors | Family history |
| Diagnostic method | Genetic testing |
| Treatment | Supportive |
| Prognosis | Varies depending on specific disorder |
| Frequency | Unknown; HSAS 1 per 30,000 male live births |
L1 syndrome can be caused by different variants in L1CAM,[3] the gene that provides the information that allows the body to produce L1 cell adhesion molecule (sometimes called the L1 protein).[3] The L1 cell adhesion molecule is a surface protein found on the surface of all neurons.[4] It allows neurons to bind to one another and create synapses (connections where information is passed on from the axons of one neuron to the dendrites and cell body of another).[4][5] As a result, L1 cell adhesion molecule is essential for the structural development of the brain and contributes to the ability to think, move, and develop memories.[4] The type and severity of L1CAM variant causing L1 syndrome in a particular person is directly related to the severity of symptoms and functional impairment that they experience.[6][7]
There is no cure for L1 syndrome, and prognosis is often poor.[8][9] Life expectancy for people with L1 syndrome can vary dramatically depending on the severity of the condition, with some dying shortly after birth and others reaching adulthood.[2] Treatment for people with L1 syndrome is supportive and aims to improve quality of life and minimize functional impairment.[10][3]
Signs and symptoms
L1 syndrome presents as a spectrum ranging from mild to severe features.[3] There is a genotype -phenotype correlation across the L1 spectrum, meaning that the specific genetic variant causing an L1-spectrum disorder in a patient determines the severity of the L1 syndrome in that patient.[6] Patients with truncating (loss-of-function) variants in L1CAM, which prevent the full synthesis of L1 (protein) experience more severe features than patients with missense variants in L1CAM, which may result in an abnormal protein but do not prevent its synthesis.[6] Illustrating this difference in L1 syndrome severity, up to 50% of infants born with L1 syndrome caused by a truncating mutation will die before the age of 3 years despite provision of best available medical treatment.[6] In comparison, roughly 10% of infants born with L1 syndrome caused by a missense mutation will die before the age of 3 years.[6]
Despite its presentation on a continuous spectrum, L1 syndrome is loosely divided into four discrete phenotypes.[11][2]
Societal Implications Based on Symptoms
People diagnosed with L1 syndrome often experience issues with respect to societal roles and interactions due to the severe physical and mental disabilities associated with the disorder. These issues can vary depending on the symptoms that manifest in a specific individual and the severity of those symptoms, which is ultimately determined based on where the individual is situated on the L1 syndrome spectrum.
Spasticity is one of the most common signs of L1 syndrome and is seen in all four major clinical phenotypes. It is characterized as the continuous contraction of certain muscles, leading to muscle stiffness which can interfere with normal movement and speech.[12] In three of the major phenotypes (except HSAS), this spasticity is presented as spastic paraplegia, where the muscles of the lower limbs are stiff and continuously contracted.[13] This spastic paraplegia often manifests itself as a gait (walking motion) disorder, specifically shuffling gait in MASA syndrome patients,[3] which acts as a source of impairment and stress due to postural instability, and leads to poor quality of life and increased mortality.[14]
Aphasia is also a common disorder, especially in people with MASA syndrome (a disorder on the L1 syndrome spectrum) and describes a range of language impairments with respect to syntax (structure), semantics (meaning), phonology (sound), morphology (structure), and/or pragmatics in language comprehension or expression.[15] People with aphasia, as well as their family members, often experience a poor quality of life due to the social isolation and depression caused by this language impairment and therefore may seek therapy to enable functional and socially relevant communication.[15] Therapy services to address aphasia in MASA syndrome patients include one-on-one sessions with a clinician, group therapy, or even computer-based therapy, which is becoming more popular given its accessibility.[15]
Intellectual disabilities also contribute to the social difficulties faced by people with L1 syndrome, and can range from mild to severe depending on the person's location on the L1 syndrome spectrum.[3] People with mild intellectual disability usually have an IQ around 50-70 (100 is the average) and are slower in all developmental areas, but they have no unusual physical characteristics and are able to blend in socially. Moderate intellectual disability characteristics include being able to maintain self-care with some support from other, travel to familiar places in the community, communicate in simple ways, and having an IQ around 35–50. Severe intellectual disability is often seen in people with X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) (on the L1 syndrome spectrum) and has several key characteristics including: the ability to understand speech but otherwise having very limited communication skills; the ability to learn daily routines and simple self-care, but need direct supervision in social settings and need family support to live in a supervised home setting.[16][17] Depending on the severity of the intellectual disability, people with L1 syndrome will have varying levels of difficulty in adapting to their social environments and may need considerable support from others to complete day-to-day tasks.
X-linked hydrocephalus with stenosis of the aqueduct of Sylvius
X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) is the most severe phenotype on the L1 spectrum and is predominantly known for its major feature: profound hydrocephalus, typically beginning before birth.[3] Due to its prenatal onset (i.e. before the bones of the skull have fused together), hydrocephalus associated with HSAS results in progressive macrocephaly (abnormal enlargement of the skull) due to markedly increased intracranial pressure.[19] The signs and symptoms of hydrocephalus can vary depending on severity and age of onset, however irritability (due to pain) and vomiting are common amongst infants with the condition.[19] Without treatment, congenital hydrocephalus can be fatal in infancy.[20] In less severe cases of untreated hydrocephalus, a child may progress beyond infancy but often experiences nausea and vomiting, missed developmental milestones (both physical and cognitive/social), diplopia (double vision), and papilledema (swelling of the optic disc) which can progress to permanent visual impairment due to increased intracranial pressure if definitive treatment is withheld.[20][21][22] Neurological damage, caused by both hydrocephalus and poor neuronal development because of defects in the L1 cell adhesion molecule, results in nearly all people with HSAS experiencing severe intellectual disability.[6]
People living with HSAS will also frequently experience spasticity,[2] a condition causing some muscles to be continuously contracted, thereby causing stiffness of the body and challenges with walking and speaking.[23] Spasticity is also known to cause difficulty in performing activities of daily living such as bathing and showering, dressing, and self-feeding.[23][24][25]
Despite HSAS frequently being considered an isolated disorder of the central nervous system, its genetic basis also causes musculoskeletal defects that result in more than half of males with HSAS displaying thumbs that are adducted (clasped, or brought inwards towards the palm).[3] Specifically, this abnormal presentation of the hand is due to congenital malformations in the extensor pollicis brevis and/or extensor pollicis longus muscle of forearm.[26]
MASA syndrome
MASA syndrome is named after its four principle features: mental retardation, adducted thumbs (clasped, or brought inwards towards the palm), shuffling gait, and aphasia (a language disability affecting the comprehension and production of speech as well as reading and writing abilities).[27][3]
Diagnosis
.jpg.webp)
A healthcare provider, usually a medical geneticist (a physician with special training in diagnosing and managing genetic disorders) can provide a clinical diagnosis of L1 syndrome by examining a patient and ordering certain imaging studies,[3] however the presence of L1 syndrome can only be confirmed when a molecular diagnosis has been made through genetic testing.[6]
Often, the diagnostic odyssey for a person with L1 syndrome begins prenatally (before they are born) when prenatal ultrasounds reveal non-specific brain abnormalities, ventriculomegaly, or a non-existent or underdeveloped corpus callosum.[6][2] Fetuses with X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) will typically have hydrocephalus severe enough to be discovered upon routine fetal ultrasound as early as 18–20 weeks gestation.[19] After birth, in the presence of suggestive features (such as macrocephaly), hydrocephalus can be confirmed with noninvasive imaging including head magnetic resonance imaging, computed tomography, or ultrasound showing ventriculomegaly, or direct measurement of intracranial pressure through invasive techniques such as lumbar puncture.[28] Further, the spasticity found in patients with HSAS can be easily demonstrated by examining the deep tendon reflexes and the extensor plantar reflex (which will both be abnormally brisk and strong due to damage to the cortex and internal capsule of the brain).[29][3]
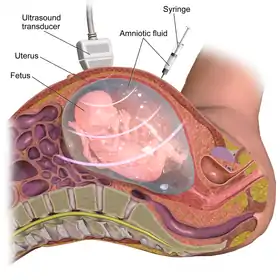
There are various types of genetic testing that can be used to confirm an L1 syndrome diagnosis in a patient.[2] After birth, genetic tests carry a low-risk of physical complications and are minimally-painful: for the patient, the process requires a small amount of blood being drawn from the arm using a needle.[32] However, prenatal genetic testing carries significant risks for both the fetus and mother due to the need to remove genetic material from the fetus while it is still in utero.[33] In order to conduct prenatal genetic testing, the mother and fetus must undergo either amniocentesis (the surgical puncturing of the amniotic sac, which holds the fetus in the womb) or, less frequently, chorionic villus sampling.[34] Amniocentesis provides a sample of amniotic fluid that can be used to screen for sequence variants or chromosomal variants,[35] whereas samples obtained through chorionic villus sampling can only be used to detect major chromosomal abnormalities (such as trisomy 21),[36] making chorionic villous sampling less relevant in the context of diagnosing L1 syndrome (which is often caused by sequence variants).[37] Amniocentesis (typically performed between 15 and 18 weeks of pregnancy) has a 1% risk of complications for mother and fetus, including miscarriage, while chorionic villus sampling (typically performed between 10 and 12 weeks of pregnancy) has a 2% risk of complications including miscarriage.[33] Mothers carrying a fetus with suspected L1 syndrome will often elect to undergo amniocentesis despite its risk, rather than waiting to pursue lower-risk genetic testing after their child is born, because prenatal diagnostic results can inform considerations to terminate the pregnancy.[38][39]
For any child born with multiple physical abnormalities, the first-line diagnostic test is chromosomal microarray.[40]
Management
To know the extent of the disease and the required needs of an individual diagnosed with L1 syndrome, some tests are recommended, including: Head imaging study, Complete neurologic evaluation, Developmental evaluation, Evaluation for Hirschsprung disease if there is a history of constipation, Consultation with a clinical geneticist and/or genetic counselor.[41] A proper management of the manifestations of the L1 syndrome involves a multidisciplinary(involving a team specialized in more than one medical fields) approach involving a team that works within these fields: pediatrics, child neurology, neurosurgery, rehabilitation, and clinical genetics.[41][42] To prevent secondary complications(conditions that occur in the course of a disease as a result), physiotherapy is recommended.[41] Prior to birth recognition of an affected fetus during pregnancy requires a plan involving multiple medical disciplines for a safe delivery for both mother and infant and to allow for an evaluation and possible treatment for hydrocephalus shortly after birth.[41]
Hydrocephalus: Surgery should be performed as needed, to shunt cerebrospinal fluid (CSF) in order to reduce pressure inside the head (intracranial pressure).[41][42] Intellectual Disability: The development of the individual should be monitored since the development outcomes are variable among affected individuals, educational programs are needed for these individuals.[41] Adducted Thumbs: Surgical procedures are generally not required, a splint may reduce the degree of the adduction, and in milder cases tendon transfer may improve the thumb function.[41][42]
Spastic paraplegia: Currently there are no specific treatments to prevent or reduce neural degeneration.[43] Treatments aim to reduce symptoms and improving balance, strength, and agility. Individuals should be evaluated periodically by a neurologist and physiatrist to evaluate progress made and to develop treatment strategies to maximize walking ability and reduce symptoms.[43] The General guidelines for follow-up and treatment can be followed while monitoring the neurologic features of the condition.[41][44] Treatment for spastic paraplegia usually involves exercise to: (1) improve and maintain cardiovascular fitness (The heart's ability to supply oxygen to the tissues) (2) reverse the reduced functional capacity (3) improve the mechanics of walking, and gait in general (4) Improve the individual's independence and sense of control.[44] Orthotics can be used to reduce the extension of the feet (toe down), which causes dragging and falling, to be noted that orthotics are usually used along with medications that decrease spasticity, such as Botox.[44]
References
- "Orphanet: MASA syndrome". www.orpha.net. Retrieved 2019-03-13.
- "L1 syndrome". Genetics Home Reference. Retrieved 2019-03-13.
- Stumpel C, Vos YJ (2015). "L1 Syndrome". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews. University of Washington, Seattle. PMID 20301657.
- "Genetics Home Reference: L1CAM gene". Genetics Home Reference. Retrieved 2019-03-20.
- Helmstaedter, Moritz; Brecht, Michael; Kevin M. Boergens; Straehle, Jakob; Gour, Anjali; Schmidt, Helene (28 September 2017). "Axonal synapse sorting in medial entorhinal cortex". Nature. 549 (7673): 469–475. Bibcode:2017Natur.549..469S. doi:10.1038/nature24005. ISSN 1476-4687. PMID 28959971. S2CID 3578786.
- Vos YJ, de Walle HE, Bos KK, Stegeman JA, Ten Berge AM, Bruining M, van Maarle MC, Elting MW, den Hollander NS, Hamel B, Fortuna AM, Sunde LE, Stolte-Dijkstra I, Schrander-Stumpel CT, Hofstra RM (March 2010). "Genotype-phenotype correlations in L1 syndrome: a guide for genetic counselling and mutation analysis" (PDF). Journal of Medical Genetics. 47 (3): 169–75. doi:10.1136/jmg.2009.071688. PMID 19846429. S2CID 206999425.
- Itoh, Kyoko; Fushiki, Shinji (2015). "The role of L1cam in murine corticogenesis, and the pathogenesis of hydrocephalus". Pathology International. 65 (2): 58–66. doi:10.1111/pin.12245. ISSN 1440-1827. PMID 25641508.
- Marín, Rosario; Ley-Martos, Miriam; Gutiérrez, Gema; Rodríguez-Sánchez, Felicidad; Arroyo, Diego; Mora-López, Francisco (2015-11-01). "Three cases with L1 syndrome and two novel mutations in the L1CAM gene". European Journal of Pediatrics. 174 (11): 1541–1544. doi:10.1007/s00431-015-2560-2. ISSN 1432-1076. PMID 25948108. S2CID 11373848.
- Chidsey, Brandalyn A.; Baldwin, Erin E.; Toydemir, Reha; Ahles, Lauren; Hanson, Heather; Stevenson, David A. (2014). "L1CAM whole gene deletion in a child with L1 syndrome". American Journal of Medical Genetics Part A. 164 (6): 1555–1558. doi:10.1002/ajmg.a.36474. ISSN 1552-4833. PMID 24668863. S2CID 21955986.
- França Jr, Marcondes C.; Lopes-Cendes, Iscia; DAbreu, Anelyssa; Martinez, Alberto R. M.; Servelhere, Katiane R.; Faber, Ingrid; França Jr, Marcondes C.; Lopes-Cendes, Iscia; D'Abreu, Anelyssa (March 2014). "Clinical features and management of hereditary spastic paraplegia". Arquivos de Neuro-Psiquiatria. 72 (3): 219–226. doi:10.1590/0004-282X20130248. ISSN 0004-282X. PMID 24676440.
- Finckh U, Schröder J, Ressler B, Veske A, Gal A (May 2000). "Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1-disease". American Journal of Medical Genetics. 92 (1): 40–6. doi:10.1002/(SICI)1096-8628(20000501)92:1<40::AID-AJMG7>3.0.CO;2-R. PMID 10797421.
- "Spasticity – Causes, Symptoms and Treatments". www.aans.org. Retrieved 2019-03-27.
- Fink, John K. (August 2003). "The hereditary spastic paraplegias: nine genes and counting". Archives of Neurology. 60 (8): 1045–1049. doi:10.1001/archneur.60.8.1045. ISSN 0003-9942. PMID 12925358.
- "Gait Disorders". www.movementdisorders.org. Retrieved 2019-03-27.
- Papathanasiou; Coppens, Patrick (2016-02-11). Aphasia and Related Neurogenic Communication Disorders. Jones & Bartlett Publishers. ISBN 9781284077315.
- The National Academies of Sciences, Engineering; Education, Division of Behavioral and Social Sciences and; Medicine, Institute of; Board on Children, Youth; Populations, Board on the Health of Select; Disorders, Committee to Evaluate the Supplemental Security Income Disability Program for Children with Mental; Wu, Joel T.; Boat, Thomas F. (2015-10-28). Clinical Characteristics of Intellectual Disabilities. National Academies Press (US).
- "Mild, Moderate, Severe Intellectual Disability Differences | HealthyPlace". www.healthyplace.com. Retrieved 2019-03-27.
- "L1 syndrome". Genetics Home Reference. Retrieved 2019-03-18.
- Kahle KT, Kulkarni AV, Limbrick DD, Warf BC (February 2016). "Hydrocephalus in children". Lancet. 387 (10020): 788–99. doi:10.1016/S0140-6736(15)60694-8. PMID 26256071. S2CID 27947722.
- Gmeiner M, Wagner H, Zacherl C, Polanski P, Auer C, van Ouwerkerk WJ, Holl K (January 2017). "Long-term mortality rates in pediatric hydrocephalus-a retrospective single-center study". Child's Nervous System. 33 (1): 101–109. doi:10.1007/s00381-016-3268-y. PMID 27766469. S2CID 34552879.
- Vinchon M, Rekate H, Kulkarni AV (August 2012). "Pediatric hydrocephalus outcomes: a review". Fluids and Barriers of the CNS. 9 (1): 18. doi:10.1186/2045-8118-9-18. PMC 3584674. PMID 22925451.
- Wang A (2018). "Papilledema". In Wang A (ed.). Emergency Neuro-ophthalmology. pp. 85–89. doi:10.1007/978-981-10-7668-8_15. ISBN 9789811076688.
{{cite book}}:|work=ignored (help) - Pandyan AD, Gregoric M, Barnes MP, Wood D, Van Wijck F, Burridge J, Hermens H, Johnson GR (January 2005). "Spasticity: clinical perceptions, neurological realities and meaningful measurement" (PDF). Disability and Rehabilitation. 27 (1–2): 2–6. doi:10.1080/09638280400014576. PMID 15799140. S2CID 7144640.
- "Spasticity – Causes, Symptoms and Treatments". www.aans.org. Retrieved 2019-03-19.
- Hardy SE (2014). "Consideration of Function & Functional Decline". In Williams BA, Chang A, Ahalt C, Chen H (eds.). Current Diagnosis & Treatment: Geriatrics (2 ed.). McGraw-Hill Education. Retrieved 2019-03-19.
- Akinleye SD, Culbertson MD, Cappelleti G, Richardson N, Choueka J (August 2018). "Intracompartmental Versus Extracompartmental Transposition of the Extensor Pollicis Longus for Treating Thumb-in-Palm Deformity: A Biomechanical Comparison". The Journal of Hand Surgery. 43 (8): 774.e1–774.e5. doi:10.1016/j.jhsa.2018.01.015. PMID 29500047. S2CID 3659987.
- "Aphasia Definitions". National Aphasia Association. Retrieved 2019-03-19.
- Volpe, Joseph J. (2017). Volpe's Neurology of the Newborn E-Book. Elsevier Health Sciences. ISBN 9780323508650. OCLC 1021807976.
- Dick, J. P. R. (2003-02-01). "The deep tendon and the abdominal reflexes". J Neurol Neurosurg Psychiatry. 74 (2): 150–153. doi:10.1136/jnnp.74.2.150. ISSN 0022-3050. PMC 1738294. PMID 12531937.
- "Amniocentesis". www.bcwomens.ca. Retrieved 2019-03-20.
- "Amniotic fluid: MedlinePlus Medical Encyclopedia". medlineplus.gov. Retrieved 2019-03-20.
- "EuroGentest: what is a genetic test?". www.eurogentest.org. Archived from the original on 2020-10-19. Retrieved 2019-03-19.
- "Public Health Agency of Canada: Genetic testing and screening". aem. 2013-02-05. Retrieved 2019-03-19.
- Alfirevic Z, Navaratnam K, Mujezinovic F (September 2017). "Amniocentesis and chorionic villus sampling for prenatal diagnosis". The Cochrane Database of Systematic Reviews. 2017 (9): CD003252. doi:10.1002/14651858.CD003252.pub2. PMC 6483702. PMID 28869276.
- "Amniocentesis". www.bcwomens.ca. Retrieved 2019-03-19.
- "Chorionic Villus Sampling". www.bcwomens.ca. Retrieved 2019-03-19.
- Alfirevic, Zarko (1999-01-25). "Early amniocentesis versus transabdominal chorion villus sampling for prenatal diagnosis". Cochrane Database of Systematic Reviews (2): CD000077. doi:10.1002/14651858.cd000077. ISSN 1465-1858. PMID 10796116. (Retracted, see doi:10.1002/14651858.cd000077)
- Veyver, Ignatia B. Van den; Eng, Christine M.; Yang, Yaping; Carter, Tiffiney G.; Nassef, Salma A.; Mathur, Veena S.; Stover, Samantha R.; Westerfield, Lauren E. (2015-10-01). "Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice". Prenatal Diagnosis. 35 (10): 1022–1029. doi:10.1002/pd.4674. ISSN 1097-0223. PMID 26275793. S2CID 206350713.
- Kohli, Jyothi Kiran (2016-11-17). "Prenatal diagnosis and screening of genetic abnormalities in early pregnancy". Journal of Evidence Based Medicine and Healthcare. 3 (92): 5053–5057. doi:10.18410/jebmh/2016/1060.
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH (May 2010). "Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies". American Journal of Human Genetics. 86 (5): 749–64. doi:10.1016/j.ajhg.2010.04.006. PMC 2869000. PMID 20466091.
- Stumpel, Connie; Vos, Yvonne J. (2015), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "L1 Syndrome", GeneReviews®, University of Washington, Seattle, PMID 20301657, retrieved 2019-03-27
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: L1 syndrome". www.orpha.net. Retrieved 2019-03-27.
- Hedera, Peter (2018), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Hereditary Spastic Paraplegia Overview", GeneReviews®, University of Washington, Seattle, PMID 20301682, retrieved 2019-03-27
- "Hereditary Spastic Paraplegia". NORD (National Organization for Rare Disorders). Retrieved 2019-03-27.