Laron syndrome
Laron syndrome (LS), also known as growth hormone insensitivity or growth hormone receptor deficiency (GHRD), is an autosomal recessive disorder characterized by a lack of insulin-like growth factor 1 (IGF-1; somatomedin-C) production in response to growth hormone (GH; hGH; somatotropin).[6] It is usually caused by inherited growth hormone receptor (GHR) mutations.[2][6]
| Laron syndrome | |
|---|---|
| Other names | genetic pituitary dwarfism (1966), Laron dwarfism (1973), Laron-type dwarfism (1984), growth hormone insensitivity (1994), hereditary somatomedin deficiency, growth hormone receptor deficiency (GHRD)(1999)[1] |
 | |
| Growth hormone | |
| Specialty | Endocrinology, Medical Genetics, Pediatrics |
| Symptoms | Short stature, truncal obesity, facial dysmorphism[2] |
| Usual onset | Present at birth |
| Duration | Lifelong |
| Causes | Autosomal recessive growth hormone receptor gene mutation (chromosome 5)[2] |
| Risk factors | Hypoglycemia, seizures, reduced intellectual capacity, osteopenia[2] |
| Differential diagnosis | STAT5b, IGF1 gene mutation, ALS deficiency, IGF-1 receptor mutation, familial short stature, malnutrition, hepatic disease, congenital growth delay, hypopituitarism[1] |
| Treatment | IGF-1, Mecasermin[3] |
| Frequency | 1–9 / 1,000,000 (approximately 250 known cases worldwide) [4][5] |
Affected individuals classically present with short stature between −4 and −10 standard deviations below median height, obesity, craniofacial abnormalities, micropenis, low blood sugar, and low serum IGF-1 despite elevated basal serum GH.[7][5][8]
LS is a very rare condition with a total of 250 known individuals worldwide.[4][5] The genetic origins of these individuals have been traced back to Mediterranean, South Asian, and Semitic ancestors, with the latter group comprising the majority of cases.[5] Molecular genetic testing for growth hormone receptor gene mutations confirms the diagnosis of LS, but clinical evaluation may include laboratory analysis of basal GH, IGF-1 and IGFBP levels, GH stimulation testing, and/or GH trial therapy. Treatment options include recombinant IGF-1 (Mecasermin).[3]
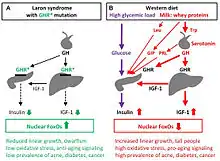
Evidence has suggested that people with Laron syndrome have a reduced risk of developing cancer and diabetes mellitus type II, with a significantly reduced incidence and delayed age of onset of these diseases compared to their unaffected relatives.[9][10] The molecular mechanisms of increased longevity and protection from age-related disease among people with LS is an area of active investigation.[11]
Presentation
Physical features
LS is recognized as being part of a spectrum of conditions that affect the Hypothalamic–pituitary–somatotropic axis and cause significant derangements in human growth, development, and metabolism.[1][6] Along this spectrum of conditions, individuals with LS and growth hormone deficiency display short stature, while individuals with acromegaly and gigantism result in the opposite phenotype of tall stature.[1][12]
In addition to short stature, other characteristic physical symptoms of LS include: prominent forehead, depressed nasal bridge, underdevelopment of mandible, truncal obesity,[7] and micropenis in males. Left untreated, the average height attained by individuals with LS are approximately 4–4.5 feet (1.2–1.4 m) in women/men respectively.[13] Additional physical features include delayed bone age, hypogonadism, blue sclera, high-pitched voice, acrohypoplasia, sparse hair growth, and crowded teeth.[14] The breasts of females reach normal size, and in some are large in relation to body size.[6] It has been suggested that hyperprolactinemia may contribute to the enlarged breast size.[9] Seizures are frequently seen secondary to hypoglycemia. Some genetic variations decrease intellectual capacity.[15] Laron syndrome patients also do not develop acne, except temporarily during treatment with IGF-1 (if performed).[9]
Pathophysiology

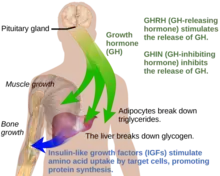
Under normal circumstances in humans, growth hormone (GH) is released in a pulsatile fashion from cells known as somatotrophs in the anterior pituitary gland.[16] These pulses of GH are regulated by cells in the hypothalamus, via the release of growth hormone-releasing hormone (GHRH) into the hypothalamohypophysial system when stimulated by insulin, ghrelin, glucagon, arginine, deep sleep, exercise, fasting, sex hormone release during puberty, and a host of other factors.[16] GH release is inhibited by somatostatin (GHIH), IGF-1, hyperglycemia, and glucocorticoids.[16] Once released, the GH molecules travel through the bloodstream and eventually bind to GH receptors on the surface of cells composing bodily organs and tissues.[16] One major site of action for GH is in the liver, where it stimulates gluconeogenesis and the release of IGF-1 through the JAK-STAT signaling pathway.[16] IGF-1 promotes growth in a variety of tissues throughout the body, especially bone mineralization, and provides negative feedback on GH release.[16] GH results in increased muscle mass, lipolysis, and protein synthesis.[16] Obesity and increased adipose tissue, especially visceral fat, results in reduced GH secretion.[16] There is a natural age-related decline in the GHRH-stimulated release of GH.[16]
Growth hormone receptor mutations
Molecular genetic investigations have shown that LS is mainly associated with autosomal recessive mutations in the gene for the growth hormone receptor (GHR).[6][17] These can result in defective hormone binding to the ectodomain or reduced efficiency of dimerization of the receptor after hormone occupancy.[18]
LS is generally classified as "primary" GH insensitivity, which is distinguished from "secondary" GH insensitivity.[1] Primary (congenital/hereditary) GH insensitivity may result from growth hormone receptor defects, as in the case of Laron syndrome, but can also be caused by defective post-receptor signal transduction (STAT5B), abnormalities of the IGF-1 gene or IGF-1 receptor.[2][1] Secondary (acquired) GH insensitivity results from antibodies to growth hormone or the growth hormone receptor, as well as poor nutritional status, liver disease or diabetes mellitus.[5][6] A GHR mutation that results in only partial insensitivity to GH can manifest as a form of idiopathic short stature.[2][8]
Diagnosis
LS should be suspected in children or adults with distinctive physical features listed above, extremely elevated serum hGH concentrations despite low serum IGF-1 levels.[14] A failure of IGF-1 to increase in response to exogenous hGH (IGF-1 stimulation test) is diagnostic for LS.[14] The gold standard for confirming a diagnosis of LS is to perform a genetic analysis with PCR to identify the precise molecular defect in the GH receptor gene.[14] Other laboratory abnormalities include GHBP (growth hormone binding protein) levels being low in cases with mutations in the extracellular domain of the GH receptor and normal in cases with mutations in the intracellular domain. Low serum levels of IGFBP are non-diagnostic for LS.[14]
Treatment
Administration of GH has no effect on IGF-1 production, therefore treatment is mainly by biosynthetic IGF-1. IGF-1 must be taken before puberty to be effective.[20]
The drug product Increlex (mecasermin), developed by the company Tercica, purchased by Ipsen, was approved by the US Food and Drug Administration in August 2005 for replacing IGF-1 in patients who are deficient.[21]
IPLEX (Mecasermin rinfabate) is composed of recombinant human IGF-1 (rhIGF-1) and its binding protein IGFBP-3. It was approved by the U.S. Food and Drug Administration (FDA) in 2005 for treatment of primary IGF-1 deficiency or GH gene deletion.[22][23] Side effects from IPLEX are hypoglycemia. IPLEX's manufacturing company, Insmed, after selling its protein production facility, can no longer develop proteins, thus can no longer manufacture IPLEX as of a statement released in July 2009.[24]
Legal battle for access to Medicine in Ecuador
In Ecuador, despite having the highest number of Laron syndrome cases in the world, affected children lacked proper treatment. In 2010 and 2014, a group of parents, led by Santiago Vasco Morales, filed lawsuits requesting the Ecuadorian government to provide the necessary comprehensive treatment. However, due to the lack of response and non-compliance with court rulings, the parents sought assistance from the Inter-American Commission on Human Rights (IACHR). Only after an admissibility report was issued by the IACHR on April 24, 2020 [25] did the Ecuadorian state begin administering the treatment. Unfortunately, many of the patients who requested treatment in 2010 were unable to benefit from it, as they reached adulthood without receiving IGF-1.
Prognosis
Cancer and diabetes
It has been reported that people with LS in Ecuador are resistant to cancer and diabetes and are somewhat protected against aging.[26][27][28] This is consistent with findings in mice with a defective growth hormone receptor gene.[20] Among the approximately 100 individuals in this population, there were no reported cases of diabetes and one case of cancer.[29]
A 2019 study of individuals with isolated growth hormone deficiency (IGHD type 1B) in Itabaianinha County, Brazil demonstrated a phenotype consistent with Laron syndrome.[30] Researchers found that these humans had similarly extended healthspan, with resistance to cancer and attenuated effects of aging, but neither patients with LS nor IGHD experienced an increase in their overall lifespan.[30]
Incidence
The majority of reported cases of Laron syndrome have been in people with Semitic origins, almost all of them being Jews or assimilated descendants of Jews.
Numerous Laron syndrome patients are found in Israel among the country's diverse Jewish population composed of Jews from around the world, as well as patients outside Israel originally from communities of the Jewish diaspora, such as Egypt and Iraq. The original "Israeli cohort" of patients referred to Zvi Laron and colleagues beginning in 1958 consisted of 64 patients as of 2009, including 4 deceased patients.[1] The countries of origin of these patients include Israel, Palestine, Jordan, Lebanon, Iran, Malta, Italy, Argentina, Ecuador, and Peru.[1]
A disproportionate number of people with the condition are found in remote villages in the Loja province of Ecuador. These individuals are descended from colonial-era Jewish-origin New Christian conversos (Sephardi Jews who themselves, or whose forebears, had been compelled to convert to Catholicism back in Spain) who had covertly migrated to Ecuador during the Spanish Conquest despite the Spanish Crown's prohibition of their emigration to its colonies and territories as a result of the Inquisition.[26][20][5][2][31]
Other patients include people of other Semitic non-Jewish origins, including from Saudi Arabia, Japan, and China.[1]
Homo floresiensis
Recent publications have proposed that Homo floresiensis represented a population with widespread Laron syndrome, based upon the many similarities of skeletal remains found in Indonesia with LS.[32][33] This is only one of several competing hypotheses, and has received criticism as insufficient to explain the "range features observed in H. floresiensis".[34] Similar postulates have been proposed regarding the Pygmies of Central Africa.[35]
History
Israeli pediatric endocrinologist Zvi Laron, along with Athalia Pertzelan, Avinoam Galatzer, Liora Kornreich, Dalia Peled, Rivka Kauli, and Beatrice Klinger published the earliest clinical studies of individuals with LS beginning in 1966.[36][37][1] Among their first 22 patients, Laron and colleagues noted consanguineous genealogy of Israeli and Palestinian ancestry with distinct physical characteristics resembling hypopituitarism.[1][6] However, researchers noticed that these people had high serum GH levels, which are expected to be low in patients with hypopituitarism.[1] Successive studies carried out over the subsequent 20 years by Laron and colleagues revealed an absence of IGF-1 release in response to exogenous hGH and an absence of GH binding to liver cell membranes in this group of patients.[1] The results of these studies provided clear evidence that the pathogenicity of the disease was the result of GH receptor failure in the liver.[1]
See also
References
- Laron Z, Kopchick J (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 3–6. ISBN 978-3-642-11183-9. Retrieved 10 November 2020.
- Hamosh A, O'Neill M, Phillips J, McKusick V. "# 262500 LARON SYNDROME". omim.org. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. Retrieved 10 November 2020.
- Grimberg A, DiVall SA, Polychronakos C (2016). "Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency". Hormone Research in Paediatrics. 86 (6): 361–397. doi:10.1159/000452150. PMID 27884013. S2CID 5798925.
- Leger J. "ORPHA:633". orpha.net. Retrieved 30 October 2020.
- Rosenbloom AL (13 November 2019). "Growth Hormone Resistance". Medscape Reference. Retrieved 3 November 2020.
- Laron Z (2004). "Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003". The Journal of Clinical Endocrinology & Metabolism. 89 (3): 1031–1044. doi:10.1210/jc.2003-031033. ISSN 0021-972X. PMID 15001582.
- Laron Z, Ginsberg S, Lilos P, Arbiv M, Vaisman N (2006). "Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity)". Clin. Endocrinol. 65 (1): 114–7. doi:10.1111/j.1365-2265.2006.02558.x. PMID 16817829. S2CID 11524548.
- Murray PG, Clayton PE (16 November 2016). Disorders of Growth Hormone in Childhood. MDText.com, Inc. PMID 25905205. Retrieved 3 November 2020.
- Laron Z, Kopchick J (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 339, 341. ISBN 978-3-642-11183-9.
- Laron Z, Kauli R, Lapkina L, Werner H (2017). "IGF-I deficiency, longevity and cancer protection of patients with Laron syndrome". Reviews in Mutation Research. 772 (123–133): 123–133. doi:10.1016/j.mrrev.2016.08.002. PMID 28528685.
- Werner H, Lapkina-Gendler L, Laron Z (2017). "Fifty years on: New lessons from the laron syndrome". Israel Medical Association Journal. 19 (1): 6–7. PMID 28457105.
- David A, Hwa V, Metherell L, Netchine I (2011). "Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity". Endocrine Reviews. 32 (4): 472–97. doi:10.1210/er.2010-0023. PMID 21525302.
- "Laron Syndrome". GARD: Genetics and Rare Disease Information Center. Retrieved 16 November 2020.
- Laron Z, Kopchick J (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 27–28. ISBN 978-3-642-11183-9. Retrieved 10 November 2020.
- Shevah O, Kornreich L, Galatzer A, Laron Z (2005). "The intellectual capacity of patients with Laron syndrome (LS) differs with various molecular defects of the growth hormone receptor gene. Correlation with CNS abnormalities". Horm. Metab. Res. 37 (12): 757–60. doi:10.1055/s-2005-921097. PMID 16372230. S2CID 260168044.
- Cohen L (4 July 2016). Growth Hormone Deficiency: Physiology and Clinical Management. Springer International Publishing. pp. 10–17. ISBN 978-3319280387. Retrieved 16 November 2020.
- Lin S, Li C, Zhang X (2018). "Growth hormone receptor mutations related to individual dwarfism". International Journal of Molecular Sciences. 19 (5): 1433. doi:10.3390/ijms19051433. PMC 5983672. PMID 29748515.
- Laron Z, Kopchick J (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 30–46. ISBN 978-3-642-11183-9. Retrieved 12 November 2020.
- Hwa V, Camacho-Hübner C, Little BM, et al. (2007). "Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene". Horm. Res. 68 (5): 218–24. doi:10.1159/000101334. PMID 17389811. S2CID 46405455.
- Wade N (17 February 2011). "Ecuadorean Villagers May Hold Secret to Longevity". The New York Times. ISSN 0362-4331. Retrieved 17 February 2011.
- "Increlex (mecasermin)". Centerwatch.com. Retrieved 21 November 2014.
- Kemp SF. "Mecasermin rinfabate". Thomson Reuters. Retrieved 5 March 2011.
- Meyer R. "Approval letter (Mecasermin rinfabate)" (PDF). FDA. Retrieved 5 March 2011.
- "Insmed Provides Update on Supply of IPLEX(TM)". Retrieved 25 August 2017.
- "Report No. 75/20 - Petition 1011-11 - Report on Admissibility" (PDF). www.oas.org. Archived from the original (PDF) on 20 October 2020. Retrieved 9 June 2023.
- Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. (2011). "Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans". Science Translational Medicine. 3 (70): 70ra13. doi:10.1126/scitranslmed.3001845. PMC 3357623. PMID 21325617.
- Bai N. "Defective Growth Gene in Rare Dwarfism Disorder Stunts Cancer and Diabetes". Scientific American. Retrieved 17 February 2011.
- Winerman L. "Study: Dwarfism Gene May Offer Protection From Cancer, Diabetes". PBS. Retrieved 17 February 2011.
- Bartke A (2012). "Healthy aging: Is smaller better? - A mini-review". Gerontology. 58 (4): 337–343. doi:10.1159/000335166. PMC 3893695. PMID 22261798.
- Aguiar-Oliveira M, Bartke A (2019). "Growth hormone deficiency: Health and longevity". Endocrine Reviews. 40 (2): 575–601. doi:10.1210/er.2018-00216. PMC 6416709. PMID 30576428.
- Rosenbloom A, Guevara Aguirre J, Rosenfeld R, Fielder P (15 November 1990). "The little women of Loja--growth hormone-receptor deficiency in an inbred population of southern Ecuador". The New England Journal of Medicine. 323 (20): 1367–1374. doi:10.1056/NEJM199011153232002. PMID 2233903. Retrieved 10 November 2020.
- Hershkovitz I, Kornreich L, Laron Z (2007). "Comparative skeletal features between Homo floresiensis and patients with primary growth hormone insensitivity (Laron syndrome)". Am. J. Phys. Anthropol. 134 (2): 198–208. doi:10.1002/ajpa.20655. PMID 17596857.
- Culotta E (2007). "Paleoanthropology. The fellowship of the hobbit". Science. 317 (5839): 740–742. doi:10.1126/science.317.5839.740. PMID 17690271. S2CID 89469580.
- Aiello LC (2010). "Five years of Homo floresiensis". American Journal of Physical Anthropology. 142 (2): 167–79. doi:10.1002/ajpa.21255. ISSN 0002-9483. PMID 20229502.
- Laron Z, Kopchick J (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 49–50. ISBN 978-3-642-11183-9. Retrieved 12 November 2020.
- synd/2825 at Who Named It?
- Laron Z, Pertzelan A, Mannheimer S (1966). "Genetic pituitary dwarfism with high serum concentration of growth hormone—a new inborn error of metabolism?". Isr. J. Med. Sci. 2 (2): 152–5. PMID 5916640.
External links
- Laron+syndrome at the U.S. National Library of Medicine Medical Subject Headings (MeSH)