Donohue syndrome
Donohue syndrome (also known as leprechaunism) is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features (small stature, bulging eyes, thick lips, and upturned nostrils) exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors.[1] As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia (high blood glucose levels), hypoglycemia (low blood glucose levels), hyperinsulemia (high blood insulin levels), and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
| Donohue syndrome | |
|---|---|
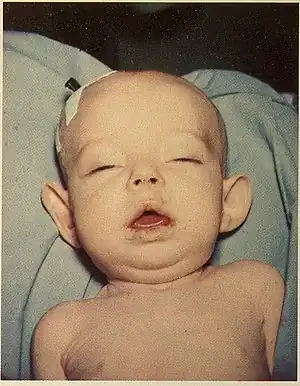 | |
| Infant with Donohue syndrome | |
| Specialty | Endocrinology, rheumatology, medical genetics |
Signs and symptoms
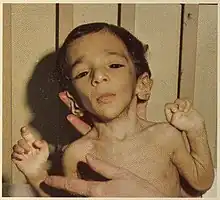
Facial features indicative of Donohue syndrome include protuberant and low-set ears, flaring nostrils, unusually large mouth, thick lips, and widely spaced eyes. Physical features include stunted growth (including during gestation), lack of subcutaneous adipose tissue, muscle atrophy, hirsutism (excessive body hair growth), and dysplasia (nail malformation).[2] Additionally, a condition known as acanthosis nigricans is present in affected individuals. In acanthosis nigricans, patches of skin darken and thicken to gain a velvet-like appearance. Gender specific features also include enlarged clitoris and breasts, as well as ovarian cysts in affected females, and enlarged penis in affected males.[2] In the Journal of Pediatric Medicine, Donohue and Uchida described affected sisters whose growth appeared to have ended in the seventh month of gestation, both born alive but dying before four months of age.[3] Very early death (or spontaneous abortion) is typical, although affected individuals sometimes live longer than a decade.[3]
Endocrine related abnormalities as a result of insulin receptor malfunction include insulin resistance, hypoglycemia and hyperglycemia (depending on whether or not the individual has eaten) and hyperinsulemia.
A much milder form of the disease, in which there is some insulin resistance but normal growth and subcutaneous fat distribution, is also known.[4] It is caused by a less severe mutation of the same gene.
Cause

Donohue syndrome is an autosomal recessive genetic disorder. The mutations responsible for the disorder are found on the short arm chromosome 19 (19p13.2) within the coding sequence of the INSR gene (insulin receptor) causing the production of inactive receptor molecules.[5] There are several mutations that can be responsible for the disease, as any mutation that severely impairs the functionality of the insulin receptor will have similar effects. The INSR gene spans over one hundred and twenty thousand base pairs, which contain twenty-two exons coding for a protein that consists of 1382 amino acids.[6] Some of the introns may or may not be spliced out depending on the kind of cell.[7]
Known mutations to the gene which can cause Donohue syndrome include a nonsense mutation that resulted in early termination of the protein, an addition or deletion mutation that resulted in a frame shift,[8] a single missense mutation[3] and in the milder form mentioned above, a single codon change that altered isoleucine to methionine in the receptor protein.[3] Some mutations to the gene instead result in insulin resistant diabetes without Donohue syndrome.[3]
Because mutations in the gene are extremely rare, most cases result from consanguineous matings, for example, between cousins.[3] However, the exact mutation need not be the same. Disease can be caused by inheritance of two different mutant alleles, one from each parent, in which case the patient is a compound heterozygote.[9]
A heterozygous individual (i.e. one who is a carrier for the disease, having only one normal allele for the insulin receptor) will not be affected. Two heterozygous parents have, in theory, a one in four chance of having a child with the disease, and two thirds of their unaffected children will be carriers. However, because spontaneous abortion (miscarriage) often results when the fetus has the disease, in actuality the proportion of children born alive with Donohue syndrome will be lower than 25%.[3]
It is possible to do a genetic test to identify carriers, but because it is so rare, this is not usually done unless there is reason to suspect that the individual being tested is a carrier, for instance having an affected sibling or cousin. As expected for a genetic disease that can be caused by many different mutations, it is not limited to a specific ethnic group, and has been seen in people of various races.
Pathophysiology
The cause of the disease is the lack of a fully functional insulin receptor, which has a profound effect during fetal development and thereafter. In one case, it was found (by culturing pancreatic cells) that the receptor produced by the mutant allele is only about 15% as effective as the normal receptor.[3] The beta cells in the pancreas, which make and store insulin and release it on an as-needed basis, are often found to be very large or numerous.[3]
The role of insulin in the body is to facilitate the entrance of glucose into the cell. Once insulin binds to the insulin receptors on the cell surface, the insulin receptors will send a signal that will ultimately bring the glucose transporter protein GLUT4 to attach to the cell membrane. Additionally, once insulin is bound to the insulin receptors, it will also initiate several signaling cascades that will promote cell growth and differentiation, protein synthesis, glucose synthesis, and the inhibition of gluconeogenesis through several metabolic pathways. A malfunctioning insulin receptor would thus not be able to properly initiate the signaling cascades for the aforementioned cellular processes. Many of the problems associated with Donohue syndrome may be due to the insulin receptor binding the insulin-like growth factor, regulating the growth of the embryo, in addition to its well-known role in the regulation of blood sugar.[10]
Diagnosis
There are a few ways to diagnose Donohue syndrome. Due to the nature of the disorder, Donohue syndrome can be diagnosed either genetically, symptomatically, or both. Because Donohue syndrome is a genetic disorder, genetic testing can be performed to diagnose the disease. These genetic tests include diagnostic testing, carrier testing, predictive and pre-symptomatic testing, as well as forensic testing. Prenatally, amniocentesis can be performed to determine if the child will have Donohue syndrome.[1] Additionally, the disorder can be diagnosed through laboratory testing to measure blood insulin levels and defective insulin receptors.
Treatment
While there currently is no cure for Donohue syndrome, treatments for those with the disease are tailored specifically to the symptoms present in each individual. It is often that a team of medical professionals will come together to treat a patient with this condition in their specific realm of practice such as pediatrics, endocrinology, and dermatology.[2] Treatment will often address specific dysfunctions in the patient, such as skin defects, hormonal imbalances, and normal progression of child growth.
Prognosis
The prognosis is quite dire, with early death usual.[5] In fact, most patients die in their first year except in milder forms of the disease, but few are known to have lived longer.[3] The variation is unsurprising given the diversity of mutations causing the disease.
Epidemiology
Donohue syndrome is an extremely rare disorder that occurs in one of every million births worldwide. Several dozen cases have been reported in the medical community, and in the reported cases of the disorder, it has been found that the females are twice as likely to have the disorder as men.
Eponym
Donohue syndrome was first identified in 1948 by Canadian pathologist William L. Donohue (1906–1985).[11] The name leprechaunism has been largely abandoned because of the perception of the name by some parents of patients as insulting.[3]
Future research
The National Institute of Diabetes and Digestive Kidney Diseases sponsored a phase 2 clinical study in 2001 that would look at the effectiveness of leptin to treat severe insulin resistance. In the study, two children with severe insulin resistance of ages 11 and 13 with known a known defect in the insulin receptor. The goal for the study was to see if leptin could overcome insulin receptor defects by initiating molecules in the insulin-signal cascade.[12] While no outcomes have yet been reported to date, the direction in which this clinical trial is heading is promising.
References
- Reference, Genetics Home. "Donohue syndrome". Genetics Home Reference. Retrieved 2019-11-19.
- "Leprechaunism". NORD (National Organization for Rare Disorders). Retrieved 2019-11-19.
- Online Mendelian Inheritance in Man (OMIM): Insulin Receptor - 147670
- al-Gazali LI, Khalil M, Devadas K (1993). "A syndrome of insulin resistance resembling leprechaunism in five sibs of consanguineous parents". J. Med. Genet. 30 (6): 470–5. doi:10.1136/jmg.30.6.470. PMC 1016418. PMID 8326490.
- Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D (2002). "Genotype-phenotype correlation in inherited severe insulin resistance". Hum. Mol. Genet. 11 (12): 1465–75. doi:10.1093/hmg/11.12.1465. PMID 12023989.
- NCBI Sequence Viewer v2.0
- Benecke H, Flier JS, Moller DE (1992). "Alternatively spliced variants of the insulin receptor protein. Expression in normal and diabetic human tissues". J. Clin. Invest. 89 (6): 2066–70. doi:10.1172/JCI115819. PMC 295926. PMID 1602013.
- Psiachou H, Mitton S, Alaghband-Zadeh J, Hone J, Taylor SI, Sinclair L (1993). "Leprechaunism and homozygous nonsense mutation in the insulin receptor gene". Lancet. 342 (8876): 924. doi:10.1016/0140-6736(93)91970-W. PMID 8105179. S2CID 8869552.
- Elsas LJ, Endo F, Strumlauf E, Elders J, Priest JH (1985). "Leprechaunism: an inherited defect in a high-affinity insulin receptor". Am. J. Hum. Genet. 37 (1): 73–88. PMC 1684537. PMID 3883764.
- Personal communication with J. Bell, Ph.D.
- Donohue WL; Edwards, HE (1948). "Dysendocrinism". The Journal of Pediatrics. 32 (6): 739–48. doi:10.1016/S0022-3476(48)80231-3. PMID 18866943.
- "Search of: "Leprechaunism" - List Results - ClinicalTrials.gov". www.clinicaltrials.gov. Retrieved 2019-11-24.