Incomplete lineage sorting
Incomplete lineage sorting,[1][2][3] also termed hemiplasy, deep coalescence, retention of ancestral polymorphism, or trans-species polymorphism, describes a phenomenon in population genetics when ancestral gene copies fail to coalesce (looking backwards in time) into a common ancestral copy until deeper than previous speciation events. It is caused by lineage sorting of genetic polymorphisms that were retained across successive nodes in the species tree.[4] In other words, the tree produced by a single gene differs from the population or species level tree, producing a discordant tree. Whatever the mechanism, the result is that a generated species level tree may differ depending on the selected genes used for assessment.[5][6] This is in contrast to complete lineage sorting, where the tree produced by the gene is the same as the population or species level tree. Both are common results in phylogenetic analysis, although it depends on the gene, organism, and sampling technique.
Concept
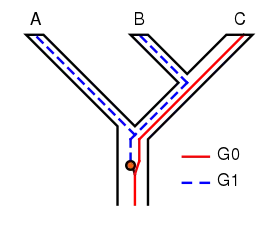
The concept of incomplete lineage sorting has some important implications for phylogenetic techniques. The persistence of polymorphisms across different speciation events can cause incomplete lineage sorting. Suppose two subsequent speciation events occur where an ancestor species gives rise firstly to species A, and secondly to species B and C. When studying a single gene, it can have multiple versions (alleles) causing different characters to appear (polymorphisms). In the example shown in Figure 1, the gene G has two versions (alleles), G0 and G1. The ancestor of A, B and C originally had only one version of gene G, G0. At some point, a mutation occurred and the ancestral population became polymorphic, with some individuals having G0 and others G1. When species A split off, it retained only G1, while the ancestor of B and C remained polymorphic. When B and C diverged, B retained only G1 and C only G0; neither were now polymorphic in G. The tree for gene G shows A and B as sisters, whereas the species tree shows B and C as sisters. If the phylogeny of these species is based on gene G, it will not represent the actual relationships between the species. In other words, the most related species will not necessarily inherit the most related genes. This is of course a simplified example of incomplete lineage sorting, and in real research it is usually more complex containing more genes and species.[7][8]
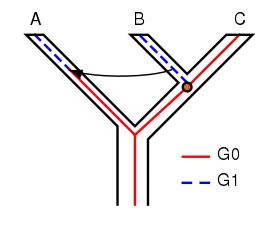
However, other mechanisms can lead to the same apparent discordancy, for example, alleles can move across species boundaries via hybridization, and DNA can be transferred between species by viruses.[9] This is illustrated in Figure 2. Here the ancestor of A, B and C, and the ancestor of B and C, had only the G0 version of gene G. A mutation occurred at the divergence of B and C, and B acquired a mutated version, G1. Some time later, the arrow shows that G1 was transferred from B to A by some means (e.g. hybridization or horizontal gene transfer). Studying only the final states of G in the three species makes it appear that A and B are sisters rather than B and C, as in Figure 1, but in Figure 2 this is not caused by incomplete lineage sorting.
Implications
Incomplete lineage sorting has important implications for phylogenetic research. There is a chance that when creating a phylogenetic tree it may not resemble actual relationships because of this incomplete lineage sorting. However, gene flow between lineages by hybridization or horizontal gene transfer may produce the same conflicting phylogenetic tree. Distinguishing these different processes may seem difficult, but much research and different statistical approaches are (being) developed to gain greater insight in these evolutionary dynamics.[10] One of the resolutions to reduce the implications of incomplete lineage sorting is to use multiple genes for creating species or population phylogenies. The more genes used, the more reliable the phylogeny becomes.[8]
In diploid organisms
Incomplete lineage sorting commonly happens with sexual reproduction because the species cannot be traced back to a single person or breeding pair. When organism tribe populations are large (i.e. thousands) each gene has some diversity and the gene tree consists of other pre-existing lineages. If the population is bigger these ancestral lineages are going to persist longer. When you get large ancestral populations together with closely timed speciation events these different pieces of DNA retain conflicting affiliations. This makes it hard to determine a common ancestor or points of branching.[5]
In primate evolution
When studying primates, chimpanzees and bonobos are more related to each other than any other taxa and are thus sister taxa. Still, for 1.6% of the bonobo genome, sequences are more closely related to homologues of humans than to chimpanzees, which is probably a result of incomplete lineage sorting.[5] A study of more than 23,000 DNA sequence alignments in the family Hominidae (great apes, including humans) showed that about 23% did not support the known sister relationship of chimpanzees and humans.[9]
In human evolution
In human evolution, incomplete lineage sorting is used to diagram hominin lineages that may have failed to sort out at the same time that speciation occurred in prehistory.[11] Due to the advent of genetic testing and genome sequencing, researchers found that the genetic relationships between hominin lineages might disagree with previous understandings of their relatedness based on physical characteristics.[11] Moreover, divergence of the last common ancestor (LCA) may not necessarily occur at the same time as speciation.[12] Lineage sorting is a method that allows paleoanthropologists to explore the genetic relationships and divergences that may not fit with their previous speciation models based on phylogeny alone.[11]
Incomplete lineage sorting of the human family tree is an area of great interest. There are a number of unknowns when considering both the transition from archaic humans to modern humans and divergence of the other great apes from the hominin lineage.[13]
Ape and hominin / human divergence
Incomplete lineage sorting means that the average divergence time between genes may differ from the divergence time between species. Models suggest that the average divergence time between the genes in the human and chimpanzee genome is older than the split between humans and gorillas. What this means is the common ancestor of humans and chimpanzees has left traces of genetic material that was present in the common ancestor of humans, chimpanzees, and gorillas.[12] However, the genetic tree slightly differs from that of the species or phylogeny tree.[14] In the phylogeny tree when we look at the evolutionary relationship between the human, bonobo chimpanzee, and gorilla, the results show that the separation of bonobo and chimpanzee transpired in a close proximity of time to the common ancestor of the bonobo-chimpanzee ancestor and humans,[12] indicating that humans and chimpanzees shared a common ancestor for several million years after separation from gorillas. This creates the phenomenon that is incomplete lineage sorting. Today researchers are relying on DNA fragments in order to study the evolutionary relationships among humans and their counterparts in the hope that it will provide information about speciation and ancestral processes from genomes from different types of humans.[15]
In viruses
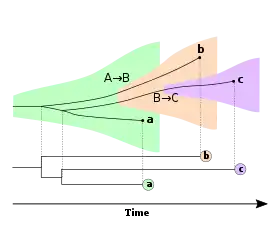
Incomplete lineage sorting is a common feature in viral phylodynamics, where the phylogeny represented by transmission of a disease from one person to the next, which is to say the population level tree, often doesn't correspond to the tree created from a genetic analysis due to the population bottlenecks that are an inherent feature of viral transmission of disease. Figure 3 illustrates how this can occur. This has relevance to criminal transmission of HIV where in some criminal cases, a phylogenetic analysis of one or two genes from the strains from the accused and the victim have been used to infer transmission; however, the commonality of incomplete lineage sorting means that transmission cannot be inferred solely on the basis of such a basic analysis.[16]
In linguistics
Jacques and List (2019)[17] show that the concept of incomplete lineage sorting can be applied to account for non-treelike phenomena in language evolution. Kalyan and François (2019), proponents of the method of historical glottometry, a model challenging the applicability of the tree model in historical linguistics, concur that "Historical Glottometry does not challenge the family tree model once incomplete lineage sorting has been taken into account."[18]
See also
References
- Simpson, Michael G (2010-07-19). Plant Systematics. Academic Press. ISBN 9780080922089.
- Kuritzin, A; Kischka, T; Schmitz, J; Churakov, G (2016). "Incomplete Lineage Sorting and Hybridization Statistics for Large-Scale Retroposon Insertion Data". PLOS Computational Biology. 12 (3): e1004812. Bibcode:2016PLSCB..12E4812K. doi:10.1371/journal.pcbi.1004812. PMC 4788455. PMID 26967525.
- Suh, A; Smeds, L; Ellegren, H (2015). "The Dynamics of Incomplete Lineage Sorting across the Ancient Adaptive Radiation of Neoavian Birds". PLOS Biology. 13 (8): e1002224. doi:10.1371/journal.pbio.1002224. PMC 4540587. PMID 26284513.
- Maddison, Wayne P. (1997-09-01). Wiens, John J. (ed.). "Gene Trees in Species Trees". Systematic Biology. Oxford University Press (OUP). 46 (3): 523–536. doi:10.1093/sysbio/46.3.523. ISSN 1076-836X.
- Rogers, Jeffrey; Gibbs, Richard A. (2014-05-01). "Comparative primate genomics: emerging patterns of genome content and dynamics". Nature Reviews Genetics. 15 (5): 347–359. doi:10.1038/nrg3707. PMC 4113315. PMID 24709753.
- Shen, Xing-Xing; Hittinger, Chris Todd; Rokas, Antonis (2017). "Contentious relationships in phylogenomic studies can be driven by a handful of genes". Nature Ecology & Evolution. 1 (5): 126. doi:10.1038/s41559-017-0126. ISSN 2397-334X. PMC 5560076. PMID 28812701.
- Copetti, Dario; Búrquez, Alberto; Bustamante, Enriquena; Charboneau, Joseph L. M.; Childs, Kevin L.; Eguiarte, Luis E.; Lee, Seunghee; Liu, Tiffany L.; McMahon, Michelle M.; Whiteman, Noah K.; Wing, Rod A.; Wojciechowski, Martin F. & Sanderson, Michael J. (2017-11-07). "Extensive gene tree discordance and hemiplasy shaped the genomes of North American columnar cacti". Proceedings of the National Academy of Sciences. 114 (45): 12003–12008. Bibcode:2017PNAS..11412003C. doi:10.1073/pnas.1706367114. PMC 5692538. PMID 29078296.
- Futuyma, Douglas J. (2013-07-15). Evolution (3rd ed.). Sunderland, Massachusetts U.S.A. ISBN 9781605351155. OCLC 824532153.
{{cite book}}: CS1 maint: location missing publisher (link) - Avise, John C. & Robinson, Terence J. (2008). "Hemiplasy: A New Term in the Lexicon of Phylogenetics". Systematic Biology. 57 (3): 503–507. doi:10.1080/10635150802164587. PMID 18570042.
- Warnow, Tandy; Bayzid, Md Shamsuzzoha; Mirarab, Siavash (2016-05-01). "Evaluating Summary Methods for Multilocus Species Tree Estimation in the Presence of Incomplete Lineage Sorting". Systematic Biology. 65 (3): 366–380. doi:10.1093/sysbio/syu063. ISSN 1063-5157. PMID 25164915.
- Maddison, Wayne P. (1997-09-01). "Gene Trees in Species Trees". Systematic Biology. 46 (3): 523–536. doi:10.1093/sysbio/46.3.523. ISSN 1076-836X.
- Mailund, Thomas; Munch, Kasper; Schierup, Mikkel Heide (2014-11-23). "Lineage Sorting in Apes". Annual Review of Genetics. 48 (1): 519–535. doi:10.1146/annurev-genet-120213-092532. ISSN 0066-4197. PMID 25251849.
- Nichols, Richard (July 2001). "Gene trees and species trees are not the same". Trends in Ecology & Evolution. 16 (7): 358–364. doi:10.1016/s0169-5347(01)02203-0. ISSN 0169-5347. PMID 11403868.
- "Primate Speciation: A Case Study of African Apes | Learn Science at Scitable". www.nature.com. Retrieved 2020-05-30.
- Peyrégne, Stéphane; Boyle, Michael James; Dannemann, Michael; Prüfer, Kay (September 2017). "Detecting ancient positive selection in humans using extended lineage sorting". Genome Research. 27 (9): 1563–1572. doi:10.1101/gr.219493.116. ISSN 1088-9051. PMC 5580715. PMID 28720580.
- Leitner, Thomas (May 2019). "Phylogenetics in HIV transmission: taking within-host diversity into account". Current Opinion in HIV and AIDS. 14 (3): 181–187. doi:10.1097/COH.0000000000000536. ISSN 1746-630X. PMC 6449181. PMID 30920395.
- Jacques, Guillaume; List, Johann-Mattis (2019). "Why we need tree models in linguistic reconstruction (and when we should apply them)". Journal of Historical Linguistics. 9 (1): 128–167. doi:10.1075/jhl.17008.mat. hdl:21.11116/0000-0004-4D2E-4. ISSN 2210-2116. S2CID 52220491.
- Kalyan, Siva; François, Alexandre (2019). "When the waves meet the trees". Journal of Historical Linguistics. 9 (1): 168–177. doi:10.1075/jhl.18019.kal. ISSN 2210-2116. S2CID 198707375.
External links
- Venema, D. (2013-08-01). "Evolution Basics: Incomplete Lineage Sorting and Ancestral Population Sizes". BioLogos. Retrieved 29 June 2018.
- Maddison, Wayne P.; Knowles, L. Lacey; Collins, Tim (2006). "Inferring Phylogeny Despite Incomplete Lineage Sorting". Systematic Biology. 55 (1): 21–30. doi:10.1080/10635150500354928. ISSN 1076-836X. PMID 16507521.
- Joly, Simon; McLenachan, Patricia A.; Lockhart, Peter J. (2009). "A Statistical Approach for Distinguishing Hybridization and Incomplete Lineage Sorting". The American Naturalist. 174 (2): E54–E70. doi:10.1086/600082. PMID 19519219. S2CID 205991354.
- Carstens, Bryan C.; Knowles, L. Lacey; Collins, Tim (2007). "Estimating Species Phylogeny from Gene-Tree Probabilities Despite Incomplete Lineage Sorting: An Example from Melanoplus Grasshoppers". Systematic Biology. 56 (3): 400–411. doi:10.1080/10635150701405560. ISSN 1076-836X. PMID 17520504. S2CID 11591644.
- Scornavacca, C.; Galtier, N. (2017). "Incomplete lineage sorting in mammalian phylogenomics". Systematic Biology. 66 (1): 112–120. doi:10.1093/sysbio/syw082. PMID 28173480.