Neuronal ceroid lipofuscinosis
Neuronal ceroid lipofuscinosis is the general name for a family of at least eight genetically separate neurodegenerative lysosomal storage diseases that result from excessive accumulation of lipopigments (lipofuscin) in the body's tissues.[1] These lipopigments are made up of fats and proteins. Their name comes from the word stem "lipo-", which is a variation on lipid, and from the term "pigment", used because the substances take on a greenish-yellow color when viewed under an ultraviolet light microscope. These lipofuscin materials build up in neuronal cells and many organs, including the liver, spleen, myocardium, and kidneys.
| Neuronal ceroid lipofuscinosis | |
|---|---|
| Other names | NCL |
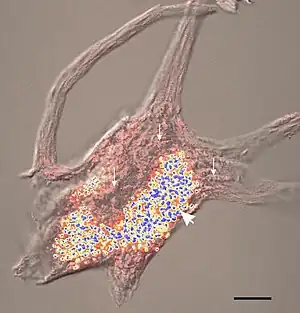 | |
| Confocal image of a spinal motor neuron showing stained lipofuscin granules in blue and yellow. | |
| Specialty | Endocrinology |
Signs and symptoms
The classic characterization of the group of neurodegenerative, lysosomal storage disorders called the neuronal ceroid lipofuscinoses (NCLs) is through the progressive, permanent loss of motor and psychological ability with a severe intracellular accumulation of lipofuscins,[2][3] with the United States and Northern European populations having slightly higher frequency with an occurrence of one in 10,000.[4] Four classic diagnoses have received the most attention from researchers and the medical field, differentiated from one another by age of symptomatic onset, duration, early-onset manifestations such as blindness or seizures, and the forms which lipofuscin accumulation takes.[2]
In the early infantile variant of NCL (also called INCL or Santavuori-Haltia), probands appear normal at birth, but early visual loss leading to complete retinal blindness by the age of 2 years is the first indicator of the disease; by 3 years of age, a vegetative state is reached, and by 4 years, isoelectric encephalograms confirm brain death. Late infantile variant usually manifests between 2 and 4 years of age with seizures and deterioration of vision. The maximum age before death for late infantile variant is 10–12 years.[5][6][7][8] Juvenile NCL (JNCL, Batten disease, or Spielmeyer-Vogt), with a prevalence of one in 100,000, usually arises between 4 and 10 years of age; the first symptoms include considerable vision loss due to retinal dystrophy, with seizures, psychological degeneration, and eventual death in the mid- to late 20s or 30s ensuing.[9] Adult variant NCL (ANCL or Kuf's disease) is less understood and generally manifests milder symptoms; however, while symptoms typically appear around 30 years of age, death usually occurs 10 years later.[1]
All the mutations that have been associated with this disease have been linked to genes involved with the neural synapses metabolism – most commonly with the reuse of vesicle proteins.
Genetics
Childhood NCLs are generally autosomal recessive disorders; that is, they occur only when a child inherits two copies of the defective gene, one from each parent. When both parents carry one defective gene, each of their children faces a one in four chance of developing NCL. At the same time, each child also faces a one in two chance of inheriting just one copy of the defective gene. Individuals who have only one defective gene are known as carriers, meaning they do not develop the disease, but they can pass the gene on to their own children. The most commonly identified mutations are in the CLN3 gene, which is located on the short arm of chromosome 16 (16p12.1). The normal function of the gene is not presently known, but results in a transmembrane protein.
Adult NCL may be inherited as an autosomal recessive (Kufs), or less often, as an autosomal dominant (Parry's) disorder. In autosomal dominant inheritance, all people who inherit a single copy of the disease gene develop the disease. As a result, no carriers of the gene are unaffected.
Many authorities refer to the NCLs collectively as Batten disease.[10]
Diagnosis
Because vision loss is often an early sign, NCL may be first suspected during an eye exam. An eye doctor can detect a loss of cells within the eye that occurs in the three childhood forms of NCL. However, because such cell loss occurs in other eye diseases, the disorder cannot be diagnosed by this sign alone. Often, an eye specialist or other physician who suspects NCL may refer the child to a neurologist, a doctor who specializes in disease of the brain and nervous system. To diagnose NCL, the neurologist needs the patient's medical history and information from various laboratory tests.
Diagnostic tests used for NCLs include:
- Skin or tissue sampling: The doctor can examine a small piece of tissue under a microscope to spot typical NCL deposits. These deposits are found in many different tissues, including skin, muscle, conjunctiva, rectal and others. Blood can also be used. These deposits take on characteristic shapes, depending on the variant under which they are said to occur: granular osmophilic deposits (GRODs) are generally characteristic of INCL, while curvilinear profiles, fingerprint profiles, and mixed-type inclusions are typically found in LINCL, JNCL, and ANCL, respectively.
- Electroencephalogram or EEG: An EEG uses special patches placed on the scalp to record electrical currents inside the brain. This helps doctors see telltale patterns in the brain's electrical activity that suggest a patient has seizures.
- Electrical studies of the eyes: These tests, which include visual-evoked responses and electroretinograms, can detect various eye problems common in childhood NCLs.
- Brain scans: Imaging can help doctors look for changes in the brain's appearance. The most commonly used imaging technique is computed tomography (CT), which uses x-rays and a computer to create a sophisticated picture of the brain's tissues and structures. A CT scan may reveal brain areas that are decaying in NCL patients. An increasingly common tool is magnetic resonance imaging, which uses a combination of magnetic fields and radio waves, instead of radiation, to create a picture of the brain.
- Enzyme assay: A recent development in diagnosis of NCL is the use of enzyme assays that look for specific missing lysosomal enzymes for infantile and late infantile versions only. This is a quick and easy diagnostic test.
Types
The older classification of NCL divided the condition into four types (CLN1, CLN2, CLN3, and CLN4) based upon age of onset, while newer classifications divide it by the associated gene.[11][12]
CLN4 (unlike CLN1, CLN2, and CLN3) has not been mapped to a specific gene.
| Type | Description | OMIM | Gene |
| Type 1 | Infantile NCL (Santavuori–Haltia disease, INCL): begins between about 6 months and 2 years of age and progresses rapidly. Affected children fail to thrive and have abnormally small heads (microcephaly). Also typical are short, sharp muscle contractions called myoclonic jerks. Initial signs of this disorder include delayed psychomotor development with progressive deterioration, other motor disorders, or seizures. The infantile form has the most rapid progression and children live into their mid-childhood years. The gene responsible for infantile NCL has been identified in some cases of juvenile/adult onset. These patients are thought to have some partial enzyme production that leads to a protracted, less severe disease course. | 256730 | PPT1 |
| Type 2 | Late infantile NCL (Jansky–Bielschowsky disease, LINCL) begins between ages 2 and 4. The typical early signs are loss of muscle coordination (ataxia) and seizures, along with progressive mental deterioration, though affected children may show mild to severe delays in speech development well before other symptoms appear. This form progresses rapidly and ends in death between ages 8 and 12. | 204500 | TPP1 |
| Type 3 | Juvenile NCL (Batten disease, JNCL) begins between the ages of 5 and 8 years of age. The typical early signs are progressive vision loss, seizures, ataxia, or clumsiness. This form progresses less rapidly and ends in death in the late teens or early 20s, although some have been known to live into their 30s. | 204200 | CLN3 |
| Type 4 | Adult NCL (Kufs disease, ANCL) generally begins before the age of 40, causes milder symptoms that progress slowly, and does not cause blindness. Although age of death is variable among affected individuals, this form does shorten life expectancy. | 204300 (AR), 162350 (AD) | CLN6[13] DNAJC5 |
| Type 5 | Finnish late infantile (Finnish late infantile variant, vLINCL) – identified in Finland | 256731 | CLN5 |
| Type 6 | Variant late infantile (late infantile variant, vLINCL) – identified in Costa Rica, South America, Portugal, the United Kingdom and other nations | 601780 | CLN6 |
| Type 7 | CLN7 | 610951 | MFSD8 |
| Type 8 | CLN8 Northern epilepsy, progressive epilepsy with mental retardation (EPMR) | 610003 | CLN8 |
| Type 8 | Turkish late infantile (Turkish late infantile variant, vLINCL) – identified in Turkey | 600143 | CLN8 |
| Type 9 | Identified in Germany and Serbia | 609055 | Unknown, but possibly regulator of dihydroceramide synthase[14] |
| Type 10 | CLN10 (congenital, cathepsin D deficiency) | 116840 | CTSD |
Infantile form
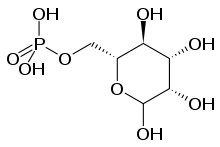
Nonsense and frameshift mutations in the CLN1 gene (located at 1p32[15][16][17]) always induce classical INCL, while some missense mutations have been associated with ANCL in addition to the infantile and juvenile forms. The mutation typically results in a deficient form of a lysosomal enzyme called palmitoyl protein thioesterase 1 (PPT1).[18]
The wild-type PPT1 is a 306-amino acid polypeptide that is typically targeted for transport into lysosomes by the mannose 6-phosphate (M6P) receptor-mediated pathway.[5][18] Here, the protein appears to function in removing palmitate residues by cleaving thioester linkages in s-acylated (or palmitoylated) proteins, encouraging their breakdown.[5][6] Defective polypeptides, however, are unable to exit the endoplasmic reticulum (ER), most likely due to misfolding; further analyses of this pathway could serve to categorize INCL among lysosomal enzyme deficiencies. The human PPT gene shows 91% similarity to bovine PPT and 85% similarity to rat PPT; these data indicate that the PPT gene is highly conserved and likely plays a vital role in cell metabolism.[5] In addition, buildup of defective PPT1 in the ER has been shown to cause the increased release of Ca2+. This homeostasis-altering event leads to increased mitochondrial membrane permeability and subsequent activation of caspase-9, eventually leading to an accumulation of cleft and uncleft poly(ADP-ribose) polymerase and eventual apoptosis.[6]
Late infantile form
The CLN2 gene encodes a 46kDa protein called lysosomal tripeptidyl peptidase I (TPP1), which cleaves tripeptides from terminal amine groups of partially unfolded proteins.[7][19] Mutations of this gene typically result in a LINCL phenotype.[20]
On April 27, 2017, the U.S. Food and Drug Administration approved cerliponase alfa (Brineura) as the first specific treatment for NCL. It is enzyme replacement therapy manufactured through recombinant DNA technology. The active ingredient in Brineura, cerliponase alfa, is intended to slow loss of walking ability in symptomatic pediatric patients 3 years of age and older with late infantile neuronal ceroid lipofuscinosis type 2 (CLN2), also known as TPP1 deficiency. Brineura is administered into the cerebrospinal fluid by infusion via a surgically implanted reservoir and catheter in the head (intraventricular access device).[21]
Juvenile form
All mutations resulting in the juvenile variant of NCL have been shown to occur at the CLN3 gene on 16p12;[16] of the mutations known to cause JNCL, 85% result from a 1.02-kb deletion, with a loss of amino acids 154–438, while the remaining 15% appear to result from either point or frameshift mutations.[9] The wild-type CLN3 gene codes for a protein with no known function,[3] but studies of the yeast CLN3 ortholog, the product of which is called battenin (after its apparent connections to Batten's disease, or JNCL), have suggested that the protein may play a role in lysosomal pH homeostasis. Furthermore, recent studies have also implied the protein's role in cathepsin D deficiency; the overexpression of the defective protein appears to have significant effects on cathepsin D processing, with implications suggesting that accumulation of ATP synthase subunit C would result.[22] Only recently have studies of human patients shown deficiency of lysosomal aspartyl proteinase cathepsin D.
Adult dominant form
Between 1.3 and 10% of cases are of the adult form. The age at onset is variable (6–62 yr). Two main clinical subtypes have been described: progressive myoclonus epilepsy (type A) and dementia with motor disturbances, such as cerebellar, extrapyramidal signs and dyskinesia (type B). Unlike the other NCLs, retinal degeneration is absent. Pathologically, the ceroid-lipofuscin accumulates mainly in neurons and contains subunit C of the mitochondrial ATP synthase.
Two independent families have been shown to have mutations in the DNAJC5 gene – one with a transversion and the other with a deletion mutation.[23] The mutations occur in a cysteine-string domain, which is required for membrane targeting/binding, palmitoylation, and oligomerization of the encoded protein cysteine-string protein alpha (CSPα). The mutations dramatically decrease the affinity of CSPα for the membrane. A second report has also located this disease to this gene.[24]
Treatment
Currently, no widely accepted treatment can cure, slow down, or halt the symptoms in the great majority of patients with NCL, but seizures may be controlled or reduced with use of antiepileptic drugs. Additionally, physical, speech, and occupational therapies may help affected patients retain functioning for as long as possible. Several experimental treatments are under investigation.
Cystagon
In 2001, a drug used to treat cystinosis, a rare genetic disease that can cause kidney failure if not treated, was reported to be useful in treating the infantile form of NCL. Preliminary results report the drug has completely cleared away storage material from the white blood cells of the first six patients, as well as slowing down the rapid neurodegeneration of infantile NCL. Currently, two drug trials are underway for infantile NCL, both using Cystagon.
Gene therapy
A gene therapy trial using an adenoassociated virus vector called AAV2CUhCLN2 began in June 2004 in an attempt to treat the manifestations of late infantile NCL.[25] The trial was conducted by Weill Medical College of Cornell University[25] and sponsored by the Nathan's Battle Foundation.[26] In May 2008, the gene therapy given to the recipients reportedly was "safe, and that, on average, it significantly slowed the disease's progression during the 18-month follow-up period"[27] and "suggested that higher doses and a better delivery system may provide greater benefit".[28]
A second gene therapy trial for late infantile NCL using an adenoassociated virus derived from the rhesus macaque (a species of Old World monkey) called AAVrh.10 began in August 2010, and is once again being conducted by Weill Medical College of Cornell University.[28] Animal models of late infantile NCL showed that the AAVrh.10 delivery system "was much more effective, giving better spread of the gene product and improving survival greatly".[28]
A third gene therapy trial, using the same AAVrh.10 delivery system, began in 2011 and has been expanded to include late infantile NCL patients with moderate tosevere impairment or uncommon genotypes, and uses a novel administration method that reduces general anesthesia time by 50% to minimize potential adverse side effects.[29]
Flupirtine
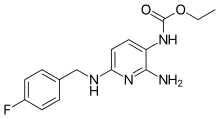
A painkiller available in several European countries, flupirtine, has been suggested to possibly slow down the progress of NCL,[30] particularly in the juvenile and late infantile forms. No trial has been officially supported in this venue, however. Currently, the drug is available to NCL families either from Germany, Duke University Medical Center in Durham, North Carolina, or the Hospital for Sick Children in Toronto.
Stem cells
On October 20, 2005, the Food and Drug Administration approved a phase-I clinical trial of neural stem cells to treat infantile and late infantile Batten disease. Subsequent approval from an independent review board also approved the stem cell therapy in early March 2006. This treatment will be the first transplant of fetal stem cells performed on humans. The therapy is being developed by Stem Cells Inc and is estimated to have six patients. The treatment was to be carried out in Oregon.[31]
Juvenile NCL has recently been listed on the Federal Clinical Trials website to test the effectiveness of bone-marrow or stem-cell transplants for this condition. A bone-marrow transplant has been attempted in the late infantile form of NCL with disappointing results; while the transplant may have slowed the onset of the disease, the child eventually developed the disease and died in 1998.
Trials testing the effectiveness of bone-marrow transplants for infantile NCL in Finland have also been disappointing, with only a slight slowing of disease reported.[32]
Immunosuppressants
In late 2007, Dr. David Pearce et al. reported that Cellcept, an immunosuppressant medication commonly used in bone-marrow transplants, may be useful in slowing down the progress of juvenile NCL.[33]
Enzyme replacement therapy
On April 27, 2017, the U.S. FDA approved cerliponase alfa as the first specific treatment for NCL.[21]
Epidemiology
Incidence can vary greatly from type-to-type, and from country-to-country.[34]
In Germany, one study reported an incidence of 1.28 per 100,000.[35]
A study in Italy reported an incidence of 0.56 per 100,000.[36]
A study in Norway reported an incidence of 3.9 per 100,000 using the years from 1978 to 1999, with a lower rate in earlier decades.[37]
History
19th century
The first probable instances of this condition were reported in 1826 in a Norwegian medical journal by Dr. Christian Stengel,[38][39][40][41] who described 4 affected siblings in a small mining community in Norway. Although no pathological studies were performed on these children the clinical descriptions are so succinct that the diagnosis of the Spielmeyer-Sjogren (juvenile) type is fully justified.
1900 to 1950
More fundamental observations were reported by F. E. Batten in 1903,[42] and by Heinrich Vogt in 1905,[43] who performed extensive clinicopathological studies on several families. Retrospectively, these papers disclose that the authors grouped together different types of the syndrome. Furthermore, Batten, at least for some time, insisted that the condition that he described was distinctly different from Tay–Sachs disease, the prototype of a neuronal lysosomal disorder now identified as GM2 gangliosidosis type A. Around the same time, Walther Spielmeyer reported detailed studies on three siblings,[44] who have the Spielmeyer-Sjogren (juvenile) type, which led him to the very firm statement that this malady is not related to Tay–Sachs disease. Subsequently, however, the pathomorphological studies of Károly Schaffer made these authors change their minds to the extent that they reclassified their respective observations as variants of Tay–Sachs disease, which caused confusion lasting about 50 years.
In 1913–14, Max Bielschowsky delineated the late infantile form of NCL.[45] However, all forms were still thought to belong in the group of "familial amaurotic idiocies", of which Tay–Sachs was the prototype.
In 1931, Torsten Sjögren, a Swedish psychiatrist and geneticist, presented 115 cases with extensive clinical and genetic documentation and came to the conclusion that the disease now called the Spielmeyer-Sjogren (juvenile) type is genetically separate from Tay–Sachs.[46]
1950 to today
Departing from the careful morphological observations of Spielmeyer, Hurst, and Sjovall and Ericsson, Zeman and Alpert made a determined effort to document the previously suggested pigmentary nature of the neuronal deposits in certain types of storage disorders.[47] Simultaneously, Terry and Korey[48] and Svennerholm[49] demonstrated a specific ultrastructure and biochemistry for Tay–Sachs disease, and these developments led to the distinct identification and also separation of the NCLs from Tay–Sachs disease by Zeman and Donahue. At that time, it was proposed that the late-infantile (Jansky–Bielschowsky), the juvenile (Spielmeyer–Vogt), and the adult form (Kufs) were quite different from Tay–Sachs disease with respect to chemical pathology and ultrastructure and also different from other forms of sphingolipidoses.
Subsequently, Santavuori and Haltia showed that an infantile form of NCL exists,[50] which Zeman and Dyken had included with the Jansky Bielschowsky type.
References
- Mole, Sara E.; Williams, Ruth E. (1 January 1993). "Neuronal Ceroid-Lipofuscinoses – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY". Neuronal Ceroid-Lipofuscinoses. PMID 20301601. Retrieved 11 December 2016.
{{cite book}}:|journal=ignored (help)update 2013 - Pardo, C.; et al. (1994). "Accumulation of the adenosine triphosphate synthase subunit c in the mnd mutant mouse". Am J Pathol. 144 (4): 829–835. PMC 1887237. PMID 8160780.
There are more than eight variants of NCL, found in 1 in 12,500 people worldwide.
- Narayan SB, Pastor JV, Mitchison HM, Bennett MJ (Aug 2004). "CLN3L, a novel protein related to the Batten disease protein, is overexpressed in Cln3-/- mice and in Batten disease". Brain. 127 (Pt 8): 1748–54. doi:10.1093/brain/awh195. PMID 15240430.
- Vesa J, Chin MH, Oelgeschläger K, et al. (Jul 2002). "Neuronal ceroid lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3". Mol Biol Cell. 13 (7): 2410–20. doi:10.1091/mbc.E02-01-0031. PMC 117323. PMID 12134079.
- Hellstein, E.; et al. (1996). "Human palmitoyl protein thioesterase". Eur Mol Bio Org J. 15 (19): 5240–5. doi:10.1002/j.1460-2075.1996.tb00909.x. PMC 452268. PMID 8895569.
- Kim, S.; et al. (2006). "PPT1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL". Hum Mol Genet. 15 (10): 1586–90. doi:10.1093/hmg/ddl078. PMID 16571600.
Late Infantile NCL (LINCL or Jansky-Bielschowsky), on the other hand, initially presents as generalized tonic-clonic or myoclonic seizures beginning at around 2–3 years of age; following this is depressed cognitive development including slow learning, speech delays, and eventual dementia leading to death, usually between 14 and 36 years of age.
- Ju, W.; et al. (2002). "Identification of CLN2 mutations shows Canadian specific NCL2 alleles". Journal of Medical Genetics. 39 (11): 822–825. doi:10.1136/jmg.39.11.822. PMC 1735024. PMID 12414822.
- Isosomppi, J.; et al. (2002). "Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein". Hum Mol Genet. 11 (8): 885–91. doi:10.1093/hmg/11.8.885. PMID 11971870.
- Persaud-Sawin, D.; et al. (2002). "Motifs within the CLN3 protein". Hum Mol Genet. 11 (18): 2129–2142. doi:10.1093/hmg/11.18.2129. PMID 12189165.
- "Batten Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 27 December 2019.
- Mole SE, Williams RE, Goebel HH (September 2005). "Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses". Neurogenetics. 6 (3): 107–26. doi:10.1007/s10048-005-0218-3. PMID 15965709. S2CID 9916771.
- Online Mendelian Inheritance in Man (OMIM): 256730
- Arsov, T; et al. (13 May 2011). "Kufs Disease, the Major Adult Form of Neuronal Ceroid Lipofuscinosis, Caused by Mutations in CLN6". American Journal of Human Genetics. 88 (5): 566–73. doi:10.1016/j.ajhg.2011.04.004. PMC 3146726. PMID 21549341.
- Schulz A, Mousallem T, Venkataramani M, et al. (February 2006). "The CLN9 protein, a regulator of dihydroceramide synthase". J. Biol. Chem. 281 (5): 2784–94. doi:10.1074/jbc.M509483200. PMID 16303764.
- NCBI Entrez Gene: PPT1 [Homo Sapiens] https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=5538&ordinalpos=28&itool=EntrezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
- Sharp, J.; et al. (1997). "Loci for classical and a variant LINCL map to chromosomes 11p15 and 15q21-23". Hum Mol Genet. 6 (4): 591–5. doi:10.1093/hmg/6.4.591. PMID 9097964.
- Online Mendelian Inheritance in Man (OMIM): NCL1, CLN1 - 256730
- Lyly, Annina; Von Schantz, C; Salonen, T; Kopra, O; Saarela, J; Jauhiainen, M; Kyttälä, A; Jalanko, A (2007). "Glycosylation, transport, and complex formation of PPT1". BMC Cell Biology. 8: 22. doi:10.1186/1471-2121-8-22. PMC 1906764. PMID 17565660.
- Gupta, P.; Hofmann, S. L. (2002). "NCL/Batten disease: the lysosomal proteinoses". Mol Psychiatry. 7 (5): 434–6. doi:10.1038/sj.mp.4001127. PMID 12082556. S2CID 5973.
Two mutations common to this gene are a G-to-C transversion and a C-to-T transition, which prematurely terminate translation at amino acid 208 of 563 (7). The deficiency of this lysosomal protease, then, results in increased subunit C storage.
- Gao, H.; et al. (2002). "Mutations in a novel CLN6-encoded transmembrane cause variant NCL in man and mouse". American Journal of Human Genetics. 70 (2): 324–35. doi:10.1086/338190. PMC 384912. PMID 11791207.
- "FDA approves first treatment for a form of Batten disease". www.fda.gov. Retrieved 30 April 2017.
- Fossale, E.; et al. (2004). "Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile ceroid lipofuscinosis". BMC Neuroscience. 5: 57. doi:10.1186/1471-2202-5-57. PMC 539297. PMID 15588329.
- Benitez BA, Alvarado D, Cai Y, Mayo K, Chakraverty S, Norton J, Morris JC, Sands MS, Goate A, et al. (2011). "Exome-sequencing confirms DNAJC5 mutations as cause of Adult Neuronal Ceroid-Lipofuscinosis". PLOS ONE. 6 (11): e26741. Bibcode:2011PLoSO...626741B. doi:10.1371/journal.pone.0026741. PMC 3208569. PMID 22073189.
- Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R, Hulkova H, Jahnova H, van der Zee J, et al. (2011). "Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis". American Journal of Human Genetics. 89 (241–252): 241–52. doi:10.1016/j.ajhg.2011.07.003. PMC 3155175. PMID 21820099.
- "Safety Study of a Gene Transfer Vector for Children With Late Infantile Neuronal Ceroid Lipofuscinosis". National Institutes of Health. Retrieved 16 December 2011.
- "Nathan's Battle – Clinical Trial Efforts". Archived from the original on 2008-05-09. Retrieved 2008-05-10.
- Klein, Andrew. "Gene therapy trial offers new hope for Batten disease, a fatal neurological disease in children". Cornell Chronicle. Retrieved May 30, 2008.
- "Safety Study of a Gene Transfer Vector (Rh.10) for Children With Late Infantile Neuronal Ceroid Lipofuscinosis". National Institutes of Health. Retrieved 16 December 2011.
- "AAVRh.10 Administered to Children With Late Infantile Neuronal Ceroid Lipofuscinosis With Uncommon Genotypes or Moderate/Severe Impairment". Retrieved 16 December 2011.
- Dhar S, Bitting RL, Rylova SN, et al. (Apr 2002). "Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons". Annals of Neurology. 51 (4): 448–66. doi:10.1002/ana.10143. PMID 11921051. S2CID 23653281.
- "Study of the Safety and Preliminary Effectiveness of Human Central Nervous System (CNS) Stem Cells (HuCNS-SC) in Patients With Infantile or Late Infantile Neuronal Ceroid Lipofuscinosis (NCL) - Full Text View - ClinicalTrials.gov". 13 January 2015.
- Lönnqvist T, Vanhanen SL, Vettenranta K, et al. (Oct 2001). "Hematopoietic stem cell transplantation in infantile neuronal ceroid lipofuscinosis". Neurology. 57 (8): 1411–6. doi:10.1212/wnl.57.8.1411. PMID 11673581. S2CID 24239827.
- "BDSRA – Batten Disease Support and Research Association". Archived from the original on 2008-07-24.
- "eMedicine – Neuronal Ceroid Lipofuscinoses : Article by Celia H Chang". 15 July 2021.
- Claussen M, Heim P, Knispel J, Goebel HH, Kohlschütter A (Feb 1992). "Incidence of neuronal ceroid-lipofuscinoses in West Germany: variation of a method for studying autosomal recessive disorders". American Journal of Medical Genetics. 42 (4): 536–8. doi:10.1002/ajmg.1320420422. PMID 1609834.
- Cardona F, Rosati E (Jun 1995). "Neuronal ceroid-lipofuscinoses in Italy: an epidemiological study". American Journal of Medical Genetics. 57 (2): 142–3. doi:10.1002/ajmg.1320570206. PMID 7668318.
- Augestad LB, Flanders WD (Nov 2006). "Occurrence of and mortality from childhood neuronal ceroid lipofuscinoses in norway". J. Child Neurol. 21 (11): 917–22. doi:10.1177/08830738060210110801. PMID 17092455. S2CID 11841986.
- Haltia M (October 2006). "The neuronal ceroid-lipofuscinoses: from past to present". Biochim. Biophys. Acta. 1762 (10): 850–6. doi:10.1016/j.bbadis.2006.06.010. PMID 16908122.
- synd/7 at Who Named It?
- C. Stengel. Beretning om et mærkeligt Sygdomstilfelde hos fire Sødskende. Eyr, 1826.
- Brean A (April 2004). "[An account of a strange instance of disease—Stengel-Batten-Spielmayer-Vogt disease]". Tidsskr. Nor. Laegeforen. (in Norwegian). 124 (7): 970–1. PMID 15088608.
- Batten, F. E. (1902). "Cerebral degeneration with symmetrical changes in the maculae in two members of a family". Transactions of the Ophthalmological Societies of the United Kingdom. 23: 386–90.
- Vogt, H. (1905). "Über familiäre amaurotische Idiotie und verwandte Krankheitsbilder". Monatsschrift für Psychiatrie und Neurologie. 18 (2): 161–71, 310–57. doi:10.1159/000213427.
- W. Spielmeyer. Klinische und anatomische Untersuchungen über eine besondere Form von familiärer amaurotische Idiotie. Freiburg im Breisgau, Gotha, 1907. Reprinted in Nissl: Histologische und histopathologische Arbeiten über die Grosshirnrinde 1908, 2: 193–213.
- Goebel HH, Gerhard L, Kominami E, Haltia M (July 1996). "Neuronal ceroid-lipofuscinosis—late-infantile or Jansky-Bielschowsky type—revisited". Brain Pathol. 6 (3): 225–8. doi:10.1111/j.1750-3639.1996.tb00850.x. PMID 8864279. S2CID 28827692.
- K. G. T. Sjögren. Die juvenile amaurotische Idiotie. 1931.
- Zeman W, Alpert M (1963). "On the nature of the "stored" lipid substances in juvenile amaurotic idiocy (Batten-Spielmeyer-Vogt)". Ann Histochim. 8: 255–7. PMID 14171739.
- Terry RD, Korey SR (Dec 1960). "Membranous cytoplasmic granules in infantile amaurotic idiocy". Nature. 188 (4755): 1000–2. Bibcode:1960Natur.188.1000T. doi:10.1038/1881000a0. PMID 13776040. S2CID 4174985.
- Svennerholm L (November 1962). "The chemical structure of normal human brain and Tay–Sachs gangliosides". Biochem. Biophys. Res. Commun. 9 (5): 436–41. doi:10.1016/0006-291X(62)90030-X. PMID 13979552.
- Santavuori P, Haltia M, Rapola J, Raitta C (Mar 1973). "Infantile type of so-called neuronal ceroid-lipofuscinosis. 1. A clinical study of 15 patients". J Neurol Sci. 18 (3): 257–67. doi:10.1016/0022-510X(73)90075-0. PMID 4698309.