Third-generation sequencing
Third-generation sequencing (also known as long-read sequencing) is a class of DNA sequencing methods currently under active development.[1]
Third generation sequencing technologies have the capability to produce substantially longer reads than second generation sequencing, also known as next-generation sequencing.[1] Such an advantage has critical implications for both genome science and the study of biology in general. However, third generation sequencing data have much higher error rates than previous technologies, which can complicate downstream genome assembly and analysis of the resulting data.[2] These technologies are undergoing active development and it is expected that there will be improvements to the high error rates. For applications that are more tolerant to error rates, such as structural variant calling, third generation sequencing has been found to outperform existing methods, even at a low depth of sequencing coverage.[3]
Current technologies
Sequencing technologies with a different approach than second-generation platforms were first described as "third-generation" in 2008–2009.[4]
There are several companies currently at the heart of third generation sequencing technology development, namely, Pacific Biosciences, Oxford Nanopore Technology, Quantapore (CA-USA), and Stratos (WA-USA). These companies are taking fundamentally different approaches to sequencing single DNA molecules.
PacBio developed the sequencing platform of single molecule real time sequencing (SMRT), based on the properties of zero-mode waveguides. Signals are in the form of fluorescent light emission from each nucleotide incorporated by a DNA polymerase bound to the bottom of the zL well.
Oxford Nanopore’s technology involves passing a DNA molecule through a nanoscale pore structure and then measuring changes in electrical field surrounding the pore; while Quantapore has a different proprietary nanopore approach. Stratos Genomics spaces out the DNA bases with polymeric inserts, "Xpandomers", to circumvent the signal to noise challenge of nanopore ssDNA reading.
Also notable is Helicos's single molecule fluorescence approach, but the company entered bankruptcy in the fall of 2015.
Advantages
Longer reads
In comparison to the current generation of sequencing technologies, third generation sequencing has the obvious advantage of producing much longer reads. It is expected that these longer read lengths will alleviate numerous computational challenges surrounding genome assembly, transcript reconstruction, and metagenomics among other important areas of modern biology and medicine.[1]
It is well known that eukaryotic genomes including primates and humans are complex and have large numbers of long repeated regions. Short reads from second generation sequencing must resort to approximative strategies in order to infer sequences over long ranges for assembly and genetic variant calling. Pair end reads have been leveraged by second generation sequencing to combat these limitations. However, exact fragment lengths of pair ends are often unknown and must also be approximated as well. By making long reads lengths possible, third generation sequencing technologies have clear advantages.
Epigenetics
Epigenetic markers are stable and potentially heritable modifications to the DNA molecule that are not in its sequence. An example is DNA methylation at CpG sites, which has been found to influence gene expression. Histone modifications are another example. The current generation of sequencing technologies rely on laboratory techniques such as ChIP-sequencing for the detection of epigenetic markers. These techniques involve tagging the DNA strand, breaking and filtering fragments that contain markers, followed by sequencing. Third generation sequencing may enable direct detection of these markers due to their distinctive signal from the other four nucleotide bases.[5]
Portability and speed

Other important advantages of third generation sequencing technologies include portability and sequencing speed.[6] Since minimal sample preprocessing is required in comparison to second generation sequencing, smaller equipments could be designed. Oxford Nanopore Technology has recently commercialized the MinION sequencer. This sequencing machine is roughly the size of a regular USB flash drive and can be used readily by connecting to a laptop. In addition, since the sequencing process is not parallelized across regions of the genome, data could be collected and analyzed in real time. These advantages of third generation sequencing may be well-suited in hospital settings where quick and on-site data collection and analysis is demanded.
Challenges
Third generation sequencing, as it currently stands, faces important challenges mainly surrounding accurate identification of nucleotide bases; error rates are still much higher compared to second generation sequencing.[2] This is generally due to instability of the molecular machinery involved. For example, in PacBio’s single molecular and real time sequencing technology, the DNA polymerase molecule becomes increasingly damaged as the sequencing process occurs.[2] Additionally, since the process happens quickly, the signals given off by individual bases may be blurred by signals from neighbouring bases. This poses a new computational challenge for deciphering the signals and consequently inferring the sequence. Methods such as Hidden Markov Models, for example, have been leveraged for this purpose with some success.[5]
On average, different individuals of the human population share about 99.9% of their genes. In other words, approximately only one out of every thousand bases would differ between any two person. The high error rates involved with third generation sequencing are inevitably problematic for the purpose of characterizing individual differences that exist between members of the same species.
Genome assembly
Genome assembly is the reconstruction of whole genome DNA sequences. This is generally done with two fundamentally different approaches.
Reference alignment
When a reference genome is available, as one is in the case of human, newly sequenced reads could simply be aligned to the reference genome in order to characterize its properties. Such reference based assembly is quick and easy but has the disadvantage of “hiding" novel sequences and large copy number variants. In addition, reference genomes do not yet exist for most organisms.
De novo assembly
De novo assembly is the alternative genome assembly approach to reference alignment. It refers to the reconstruction of whole genome sequences entirely from raw sequence reads. This method would be chosen when there is no reference genome, when the species of the given organism is unknown as in metagenomics, or when there exist genetic variants of interest that may not be detected by reference genome alignment.
Given the short reads produced by the current generation of sequencing technologies, de novo assembly is a major computational problem. It is normally approached by an iterative process of finding and connecting sequence reads with sensible overlaps. Various computational and statistical techniques, such as de bruijn graphs and overlap layout consensus graphs, have been leveraged to solve this problem. Nonetheless, due to the highly repetitive nature of eukaryotic genomes, accurate and complete reconstruction of genome sequences in de novo assembly remains challenging. Pair end reads have been posed as a possible solution, though exact fragment lengths are often unknown and must be approximated.[7]
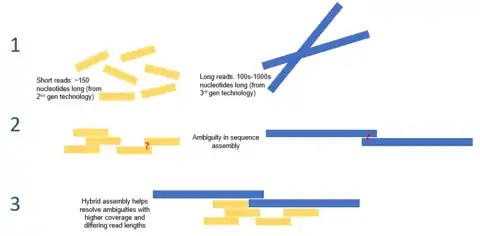
Hybrid assembly
Long read lengths offered by third generation sequencing may alleviate many of the challenges currently faced by de novo genome assemblies. For example, if an entire repetitive region can be sequenced unambiguously in a single read, no computation inference would be required. Computational methods have been proposed to alleviate the issue of high error rates. For example, in one study, it was demonstrated that de novo assembly of a microbial genome using PacBio sequencing alone performed superior to that of second generation sequencing.[8]
Third generation sequencing may also be used in conjunction with second generation sequencing. This approach is often referred to as hybrid sequencing. For example, long reads from third generation sequencing may be used to resolve ambiguities that exist in genomes previously assembled using second generation sequencing. On the other hand, short second generation reads have been used to correct errors in that exist in the long third generation reads. In general, this hybrid approach has been shown to improve de novo genome assemblies significantly.[9]
Epigenetic markers
DNA methylation (DNAm) – the covalent modification of DNA at CpG sites resulting in attached methyl groups – is the best understood component of epigenetic machinery. DNA modifications and resulting gene expression can vary across cell types, temporal development, with genetic ancestry, can change due to environmental stimuli and are heritable. After the discovery of DNAm, researchers have also found its correlation to diseases like cancer and autism.[10] In this disease etiology context DNAm is an important avenue of further research.
Advantages
The current most common methods for examining methylation state require an assay that fragments DNA before standard second generation sequencing on the Illumina platform. As a result of short read length, information regarding the longer patterns of methylation are lost.[5] Third generation sequencing technologies offer the capability for single molecule real-time sequencing of longer reads, and detection of DNA modification without the aforementioned assay.[11]
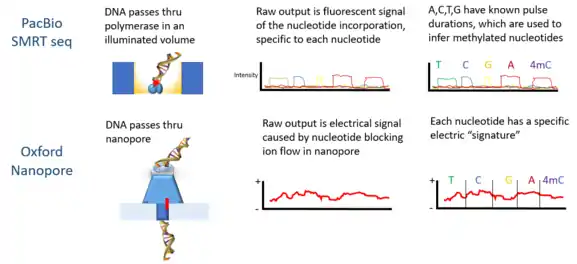
Oxford Nanopore Technologies’ MinION has been used to detect DNAm. As each DNA strand passes through a pore, it produces electrical signals which have been found to be sensitive to epigenetic changes in the nucleotides, and a hidden Markov model (HMM) was used to analyze MinION data to detect 5-methylcytosine (5mC) DNA modification.[5] The model was trained using synthetically methylated E. coli DNA and the resulting signals measured by the nanopore technology. Then the trained model was used to detect 5mC in MinION genomic reads from a human cell line which already had a reference methylome. The classifier has 82% accuracy in randomly sampled singleton sites, which increases to 95% when more stringent thresholds are applied.[5]
Other methods address different types of DNA modifications using the MinION platform. Stoiber et al. examined 4-methylcytosine (4mC) and 6-methyladenine (6mA), along with 5mC, and also created software to directly visualize the raw MinION data in a human-friendly way.[12] Here they found that in E. coli, which has a known methylome, event windows of 5 base pairs long can be used to divide and statistically analyze the raw MinION electrical signals. A straightforward Mann-Whitney U test can detect modified portions of the E. coli sequence, as well as further split the modifications into 4mC, 6mA or 5mC regions.[12]
It seems likely that in the future, MinION raw data will be used to detect many different epigenetic marks in DNA.
PacBio sequencing has also been used to detect DNA methylation. In this platform, the pulse width – the width of a fluorescent light pulse – corresponds to a specific base. In 2010 it was shown that the interpulse distance in control and methylated samples are different, and there is a "signature" pulse width for each methylation type.[11] In 2012 using the PacBio platform the binding sites of DNA methyltransferases were characterized.[13] The detection of N6-methylation in C Elegans was shown in 2015.[14] DNA methylation on N6-adenine using the PacBio platform in mouse embryonic stem cells was shown in 2016.[15]
Other forms of DNA modifications – from heavy metals, oxidation, or UV damage – are also possible avenues of research using Oxford Nanopore and PacBio third generation sequencing.
Drawbacks
Processing of the raw data – such as normalization to the median signal – was needed on MinION raw data, reducing real-time capability of the technology.[12] Consistency of the electrical signals is still an issue, making it difficult to accurately call a nucleotide. MinION has low throughput; since multiple overlapping reads are hard to obtain, this further leads to accuracy problems of downstream DNA modification detection. Both the hidden Markov model and statistical methods used with MinION raw data require repeated observations of DNA modifications for detection, meaning that individual modified nucleotides need to be consistently present in multiple copies of the genome, e.g. in multiple cells or plasmids in the sample.
For the PacBio platform, too, depending on what methylation you expect to find, coverage needs can vary. As of March 2017, other epigenetic factors like histone modifications have not been discoverable using third-generation technologies. Longer patterns of methylation are often lost because smaller contigs still need to be assembled.
Transcriptomics
Transcriptomics is the study of the transcriptome, usually by characterizing the relative abundances of messenger RNA molecules in the tissue under study. According to the central dogma of molecular biology, genetic information flows from double stranded DNA molecules to single stranded mRNA molecules where they can be readily translated into function protein molecules. By studying the transcriptome, one can gain valuable insight into the regulation of gene expressions.
While expression levels as the gene level can be more or less accurately depicted by second generation sequencing, transcript-level information still remains an important challenge.[16] As a consequence, the role of alternative splicing in molecular biology remains largely elusive. Third generation sequencing technologies hold promising prospects in resolving this issue by enabling sequencing of mRNA molecules at their full lengths.
Alternative splicing
Alternative splicing (AS) is the process by which a single gene may give rise to multiple distinct mRNA transcripts and consequently different protein translations.[17] Some evidence suggests that AS is a ubiquitous phenomenon and may play a key role in determining the phenotypes of organisms, especially in complex eukaryotes; all eukaryotes contain genes consisting of introns that may undergo AS. In particular, it has been estimated that AS occurs in 95% of all human multi-exon genes.[18] AS has undeniable potential to influence myriad biological processes. Advancing knowledge in this area has critical implications for the study of biology in general.
Transcript reconstruction
The current generation of sequencing technologies produce only short reads, putting tremendous limitation on the ability to detect distinct transcripts; short reads must be reverse engineered into original transcripts that could have given rise to the resulting read observations.[19] This task is further complicated by the highly variable expression levels across transcripts, and consequently variable read coverages across the sequence of the gene.[19] In addition, exons may be shared among individual transcripts, rendering unambiguous inferences essentially impossible.[17] Existing computational methods make inferences based on the accumulation of short reads at various sequence locations often by making simplifying assumptions.[19] Cufflinks takes a parsimonious approach, seeking to explain all the reads with the fewest possible number of transcripts.[20] On the other hand, StringTie attempts to simultaneously estimate transcript abundances while assembling the reads.[19] These methods, while reasonable, may not always identify real transcripts.
A study published in 2008 surveyed 25 different existing transcript reconstruction protocols.[16] Its evidence suggested that existing methods are generally weak in assembling transcripts, though the ability to detect individual exons are relatively intact.[16] According to the estimates, average sensitivity to detect exons across the 25 protocols is 80% for Caenorhabditis elegans genes.[16] In comparison, transcript identification sensitivity decreases to 65%. For human, the study reported an exon detection sensitivity averaging to 69% and transcript detection sensitivity had an average of a mere 33%.[16] In other words, for human, existing methods are able to identify less than half of all existing transcript.
Third generation sequencing technologies have demonstrated promising prospects in solving the problem of transcript detection as well as mRNA abundance estimation at the level of transcripts. While error rates remain high, third generation sequencing technologies have the capability to produce much longer read lengths.[21] Pacific Bioscience has introduced the iso-seq platform, proposing to sequence mRNA molecules at their full lengths.[21] It is anticipated that Oxford Nanopore will put forth similar technologies. The trouble with higher error rates may be alleviated by supplementary high quality short reads. This approach has been previously tested and reported to reduce the error rate by more than 3 folds.[22]
Metagenomics
Metagenomics is the analysis of genetic material recovered directly from environmental samples.
Advantages
The main advantage for third-generation sequencing technologies in metagenomics is their speed of sequencing in comparison to second generation techniques. Speed of sequencing is important for example in the clinical setting (i.e. pathogen identification), to allow for efficient diagnosis and timely clinical actions.
Oxford Nanopore's MinION was used in 2015 for real-time metagenomic detection of pathogens in complex, high-background clinical samples. The first Ebola virus (EBOV) read was sequenced 44 seconds after data acquisition.[23] There was uniform mapping of reads to genome; at least one read mapped to >88% of the genome. The relatively long reads allowed for sequencing of a near-complete viral genome to high accuracy (97–99% identity) directly from a primary clinical sample.[23]
A common phylogenetic marker for microbial community diversity studies is the 16S ribosomal RNA gene. Both MinION and PacBio's SMRT platform have been used to sequence this gene.[24][25] In this context the PacBio error rate was comparable to that of shorter reads from 454 and Illumina's MiSeq sequencing platforms.
Drawbacks
MinION's high error rate (~10-40%) prevented identification of antimicrobial resistance markers, for which single nucleotide resolution is necessary. For the same reason, eukaryotic pathogens were not identified.[23] Ease of carryover contamination when re-using the same flow cell (standard wash protocols don’t work) is also a concern. Unique barcodes may allow for more multiplexing. Furthermore, performing accurate species identification for bacteria, fungi and parasites is very difficult, as they share a larger portion of the genome, and some only differ by <5%.
The per base sequencing cost is still significantly more than that of MiSeq. However, the prospect of supplementing reference databases with full-length sequences from organisms below the limit of detection from the Sanger approach;[24] this could possibly greatly help the identification of organisms in metagenomics.
References
- Bleidorn, Christoph (2016-01-02). "Third generation sequencing: technology and its potential impact on evolutionary biodiversity research". Systematics and Biodiversity. 14 (1): 1–8. doi:10.1080/14772000.2015.1099575. ISSN 1477-2000. S2CID 85991118.
- Gupta, Pushpendra K. (2008-11-01). "Single-molecule DNA sequencing technologies for future genomics research". Trends in Biotechnology. 26 (11): 602–611. doi:10.1016/j.tibtech.2008.07.003. PMID 18722683.
- Tham, Cheng Yong (2020-03-03). "NanoVar: accurate characterization of patients' genomic structural variants using low-depth nanopore sequencing". Genome Biology. 21 (Article number: 56): 56. doi:10.1186/s13059-020-01968-7. PMC 7055087. PMID 32127024.
- Check Hayden, Erika (2009-02-06). "Genome sequencing: the third generation". Nature News. 457 (7231): 768–769. doi:10.1038/news.2009.86. PMID 19212365.
- Simpson, Jared T.; Workman, Rachael; Zuzarte, Philip C.; David, Matei; Dursi, Lewis Jonathan; Timp, Winston (2016-04-04). "Detecting DNA Methylation using the Oxford Nanopore Technologies MinION sequencer". bioRxiv 10.1101/047142.
- Schadt, E. E.; Turner, S.; Kasarskis, A. (2010-10-15). "A window into third-generation sequencing". Human Molecular Genetics. 19 (R2): R227–R240. doi:10.1093/hmg/ddq416. ISSN 0964-6906. PMID 20858600.
- Li, Ruiqiang; Zhu, Hongmei; Ruan, Jue; Qian, Wubin; Fang, Xiaodong; Shi, Zhongbin; Li, Yingrui; Li, Shengting; Shan, Gao (2010-02-01). "De novo assembly of human genomes with massively parallel short read sequencing". Genome Research. 20 (2): 265–272. doi:10.1101/gr.097261.109. ISSN 1088-9051. PMC 2813482. PMID 20019144.
- Chin, Chen-Shan; Alexander, David H.; Marks, Patrick; Klammer, Aaron A.; Drake, James; Heiner, Cheryl; Clum, Alicia; Copeland, Alex; Huddleston, John (2013-06-01). "Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data". Nature Methods. 10 (6): 563–569. doi:10.1038/nmeth.2474. ISSN 1548-7091. PMID 23644548. S2CID 205421576.
- Goodwin, Sara; Gurtowski, James; Ethe-Sayers, Scott; Deshpande, Panchajanya; Schatz, Michael C.; McCombie, W. Richard (2015-11-01). "Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome". Genome Research. 25 (11): 1750–1756. doi:10.1101/gr.191395.115. ISSN 1088-9051. PMC 4617970. PMID 26447147.
- Fraser, Hunter B.; Lam, Lucia L.; Neumann, Sarah M.; Kobor, Michael S. (2012-02-09). "Population-specificity of human DNA methylation". Genome Biology. 13 (2): R8. doi:10.1186/gb-2012-13-2-r8. ISSN 1474-760X. PMC 3334571. PMID 22322129.
- Flusberg, Benjamin A.; Webster, Dale R.; Lee, Jessica H.; Travers, Kevin J.; Olivares, Eric C.; Clark, Tyson A.; Korlach, Jonas; Turner, Stephen W. (2010-06-01). "Direct detection of DNA methylation during single-molecule, real-time sequencing". Nature Methods. 7 (6): 461–465. doi:10.1038/nmeth.1459. PMC 2879396. PMID 20453866.
- Stoiber, Marcus H.; Quick, Joshua; Egan, Rob; Lee, Ji Eun; Celniker, Susan E.; Neely, Robert; Loman, Nicholas; Pennacchio, Len; Brown, James B. (2016-12-15). "De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing". bioRxiv 10.1101/094672.
- Clark, T. A.; Murray, I. A.; Morgan, R. D.; Kislyuk, A. O.; Spittle, K. E.; Boitano, M.; Fomenkov, A.; Roberts, R. J.; Korlach, J. (2012-02-01). "Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing". Nucleic Acids Research. 40 (4): e29. doi:10.1093/nar/gkr1146. ISSN 0305-1048. PMC 3287169. PMID 22156058.
- Greer, Eric Lieberman; Blanco, Mario Andres; Gu, Lei; Sendinc, Erdem; Liu, Jianzhao; Aristizábal-Corrales, David; Hsu, Chih-Hung; Aravind, L.; He, Chuan (2015). "DNA Methylation on N6-Adenine in C. elegans". Cell. 161 (4): 868–878. doi:10.1016/j.cell.2015.04.005. PMC 4427530. PMID 25936839.
- Wu, Tao P.; Wang, Tao; Seetin, Matthew G.; Lai, Yongquan; Zhu, Shijia; Lin, Kaixuan; Liu, Yifei; Byrum, Stephanie D.; Mackintosh, Samuel G. (2016-04-21). "DNA methylation on N6-adenine in mammalian embryonic stem cells". Nature. 532 (7599): 329–333. Bibcode:2016Natur.532..329W. doi:10.1038/nature17640. ISSN 0028-0836. PMC 4977844. PMID 27027282.
- Steijger, Tamara; Abril, Josep F.; Engström, Pär G.; Kokocinski, Felix; The RGASP Consortium; Hubbard, Tim J.; Guigó, Roderic; Harrow, Jennifer; Bertone, Paul (2013-12-01). "Assessment of transcript reconstruction methods for RNA-seq". Nature Methods. 10 (12): 1177–1184. doi:10.1038/nmeth.2714. ISSN 1548-7091. PMC 3851240. PMID 24185837.
- Graveley, Brenton R. (2001). "Alternative splicing: increasing diversity in the proteomic world". Trends in Genetics. 17 (2): 100–107. doi:10.1016/s0168-9525(00)02176-4. PMID 11173120.
- Pan, Qun; Shai, Ofer; Lee, Leo J.; Frey, Brendan J.; Blencowe, Benjamin J. (2008-12-01). "Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing". Nature Genetics. 40 (12): 1413–1415. doi:10.1038/ng.259. ISSN 1061-4036. PMID 18978789. S2CID 9228930.
- Pertea, Mihaela; Pertea, Geo M.; Antonescu, Corina M.; Chang, Tsung-Cheng; Mendell, Joshua T.; Salzberg, Steven L. (2015-03-01). "StringTie enables improved reconstruction of a transcriptome from RNA-seq reads". Nature Biotechnology. 33 (3): 290–295. doi:10.1038/nbt.3122. ISSN 1087-0156. PMC 4643835. PMID 25690850.
- Trapnell, Cole; Williams, Brian A.; Pertea, Geo; Mortazavi, Ali; Kwan, Gordon; van Baren, Marijke J.; Salzberg, Steven L.; Wold, Barbara J.; Pachter, Lior (2010-05-01). "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation". Nature Biotechnology. 28 (5): 511–515. doi:10.1038/nbt.1621. ISSN 1087-0156. PMC 3146043. PMID 20436464.
- Abdel-Ghany, Salah E.; Hamilton, Michael; Jacobi, Jennifer L.; Ngam, Peter; Devitt, Nicholas; Schilkey, Faye; Ben-Hur, Asa; Reddy, Anireddy S. N. (2016-06-24). "A survey of the sorghum transcriptome using single-molecule long reads". Nature Communications. 7: 11706. Bibcode:2016NatCo...711706A. doi:10.1038/ncomms11706. ISSN 2041-1723. PMC 4931028. PMID 27339290.
- Au, Kin Fai; Underwood, Jason G.; Lee, Lawrence; Wong, Wing Hung (2012-10-04). "Improving PacBio Long Read Accuracy by Short Read Alignment". PLOS ONE. 7 (10): e46679. Bibcode:2012PLoSO...746679A. doi:10.1371/journal.pone.0046679. ISSN 1932-6203. PMC 3464235. PMID 23056399.
- Greninger, Alexander L.; Naccache, Samia N.; Federman, Scot; Yu, Guixia; Mbala, Placide; Bres, Vanessa; Stryke, Doug; Bouquet, Jerome; Somasekar, Sneha (2015-01-01). "Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis". Genome Medicine. 7: 99. doi:10.1186/s13073-015-0220-9. ISSN 1756-994X. PMC 4587849. PMID 26416663.
- Schloss, Patrick D.; Jenior, Matthew L.; Koumpouras, Charles C.; Westcott, Sarah L.; Highlander, Sarah K. (2016-01-01). "Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system". PeerJ. 4: e1869. doi:10.7717/peerj.1869. PMC 4824876. PMID 27069806.
- Benítez-Páez, Alfonso; Portune, Kevin J.; Sanz, Yolanda (2016-01-01). "Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION™ portable nanopore sequencer". GigaScience. 5: 4. doi:10.1186/s13742-016-0111-z. ISSN 2047-217X. PMC 4730766. PMID 26823973.
External liks
- Marx, Vivien (12 January 2023). "Method of the year: long-read sequencing". Nature Methods. 20 (1): 6–11. doi:10.1038/s41592-022-01730-w. ISSN 1548-7105.