Acute myeloblastic leukemia with maturation
Acute myeloblastic leukemia with maturation (M2) is a subtype of acute myeloid leukemia (AML).[1]
| Acute myeloblastic leukemia with maturation | |
|---|---|
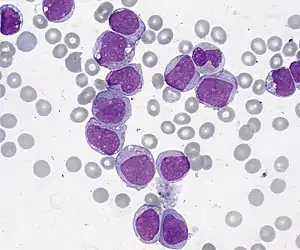 | |
| Acute Myelogenous Leukemia with maturation. Bone marrow smear, Giemsa stain, 1000x. | |
| Specialty | Hematology, oncology |
Acute myeloid leukemia (AML) is a type of cancer affecting blood cells that eventually develop into non-lymphocyte white blood cells. The disease originates from the bone marrow, the soft inner portion of select bones where blood stem cells develop into either lymphocyte or in this particular condition, myeloid cells. This acute disease prevents bone marrow cells from properly maturing, thus causing an accumulation of immature myeloblast cells in the bone marrow.
Acute myeloid leukemia is more lethal than chronic myeloid leukemia, a disease that affects the same myeloid cells, but at a different pace. Many of the immature blast cells in acute myeloid leukemia have a higher loss of function and thus, a higher inability to carry out normal functions than those more developed immature myeloblast cells in chronic myeloid leukemia (O’Donnell et al. 2012). Acute in acute myeloid leukemia means that the amounts of blast cells are increasing at a very high rate. Myeloid refers to the type of white blood cells that are affected by the condition.
Acute myeloid leukemia is the most common acute leukemia that is affecting the adult population. The 5-year survival rate for the cancer stands at around 26% (ACS, 2016).
M2 acute myeloblastic leukemia with maturation refers to the subtype of acute myeloid leukemia characterized by the maturation stages of the myeloid cell development and the location of the AML1 gene. One of the hallmarks of M2 subtype acute myeloid leukemia is the formation of a fusion protein, AML1-ETO or RUNX1-RUNX1T1, due to a translocation of chromosome 8 to chromosome 21 or t(8;21) (Miyoshi et al., 1991, Andrieu et al., 1996). This cytogenetic abnormality has been found in 90% of M2 acute myeloblastic leukemia; while the other 10% constitutes a mix of M1 and M4 acute myeloid leukemia (GFHC, 1990).
Another translocation between chromosome 6p23 and chromosome 9q34 is also associated with the M2 subtype. The t(6;9) causes the formation of a fusion oncogene made of DEK (6p23) and CAN/NUP214 (9q34). This rare translocation has a poor prognosis compared to the t(8;21) because 70% of t(6;9) acute myeloid leukemia patients have the FLT3-ITD mutation (Schwartz et al., 1983, Kottaridis, 2001). The FLT-ITD mutation is one of the most lethal mutations in acute myeloid leukemia (Chi et al., 2008).
M2 acute myeloblastic leukemia with maturation, as classified by the FAB system, constitutes 25% of adult AML.
Cause
This subtype is characterized by a translocation of a part of chromosome 8 to chromosome 21, written as t(8;21).[2] On both sides of the splice the DNA coded for different proteins, RUNX1 and ETO, These two sequences are then transcribed and translated into a single large protein, "M2 AML" which allows the cell to divide unchecked, leading to cancer.
Genetics
Acute myeloid leukemia is a very heterogeneous disease, composed of a variety of translocations and mutations. However, one tenth of all acute myeloid leukemia cases diagnosed have the AML1-ETO fusion oncoprotein due to the t(8;21) translocation. AML1 or RUNX1 is a DNA-binding transcription factor located at the 21q22. ETO is a protein with transcriptional repressing abilities located at the 8q22.
Less than 1% of acute myeloid leukemia patients have the t(6;9) mutation. The rare translocation causes the formation of fusion oncoprotein DEK-NUP214 (Huret, 2005). DEK functions as a transcriptional repressor by interfering with histone acetyl transferases, regulator for a number of stem cells, and activates gene expression in myeloid cells (Koleva et al., 2012). The NUP214 protein is involved in mRNA export as well as nuclear membrane localization and nuclear pore complex (Koser et al., 2011).
Molecular mechanism
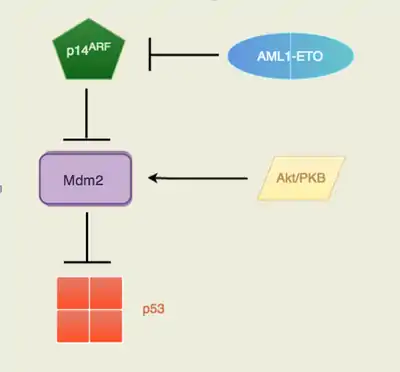
The fusion oncoprotein involves the gene AML1 (now known as RUNX1) and ETO (now known as RUNX1T1). AML1, located at the 21q22, normally has the ability to activate transcription of the ARF gene and ETO, located at the 8q22, normally has the ability to repress transcription. The fusion protein AML1-ETO is commonly found in acute myeloid leukemia patients. p14ARF is a well known tumor suppressor that serves as the safety net when p53 tumor suppressor's functions are inhibited. Many cancers recognize the potential of the p14ARF tumor suppressor to block cell growth so it is commonly mutated or inhibited in cancer cells. The AML1-ETO is incapable of p14ARF transcription as the fusion protein took on AML1's involvement with ARF gene expression and ETO's transcription repression. The Akt/PKB signaling is a pathway that is pro-survival and growth. By activating Mdm2, the signal transduction pathway will trigger the anti-apoptotic downstream effects of Mdm2. With no p14ARF to regulate and inhibit Mdm2, there will be an increased level of suppression of p53. Mdm2 is a proto-oncogene that directly antagonizes p53 to ubiquitination (Figure 1). The p53 protein is known as the “guardian of the genome” due to its ability to induce DNA repair enzymes and regulate cell cycle advancements. The down regulation of p53 by Mdm2 would lead to unchecked proliferative growth. The direct consequence of having the fusion protein, AML1-ETO, is the lack of p53 regulation in pre-leukemic cells. Therefore, there are an increased number of immature cells that are unable to carry out normal function, which is essentially cancer (Faderi et al., 2000, Song et al. 2005, Weinberg, 2014).
Autophagy in M2 AML
Autophagy is an innate pathway used for degradation of cellular components (Kobayashi, 2015). In recent studies, scientists recognize the significance of autophagy both as a potential anti-apoptotic response to cancer treatments as well as a potential mechanism for getting rid of undesirable fusion proteins such as AML1-ETO. In a 2013 study, scientists demonstrated that the degradation of fusion oncoprotein AML1-ETO is not mediated by autophagy through a set of drug dosage trials testing the levels of AML1-ETO protein expression. The acute myeloid leukemia Kasumi-1 cell line was selected for the experiment due to its AML1-ETO positive characteristics. These cells were treated with increasing concentrations of each histone deacetylase inhibitors – valproic acid (VPA) (epileptic and bipolar drug) or vorinostat (cutaneous T cell lymphoma drug), which are known to induce autophagy associated with loss of the fusion protein. The two inhibitors were added to the cell line in doses of 0, 0.38 uM, 0.74 uM, and 1.5 uM. The cell lysates were then treated with autophagy inhibitors Baf or CQ, or control. Through immunoblotting, there is no reduction of AML1-ETO observed across the different concentrations of VPA or vorinostat. The results indicate that AML1-ETO degradation is not mediated by autophagy, but there is an observed pro-survival autophagy in the leukemic cells (Torgersen et al., 2013). Thus, an inhibition of autophagy would be a viable treatment method for subtype M2 acute myeloid leukemia.
Diagnosis
The first red flag that indicates M2 acute myeloblastic leukemia with maturation is the skewed ratio of white blood cells to red blood cells. Leukemia is initially diagnosed by a peripheral blood smear, a procedure used to check for cell count and cell shapes. Then a bone marrow aspiration and biopsy would be conducted to collect and view the bone, bone marrow, and blood under a microscope. Cytogenetic assays, such as fluorescence in situ hybridization (FISH) would help evaluate the structure and function of the cell's chromosomes.
The criteria for an acute myeloid leukemia case to fall under the M2 subtype is the following: 20%+ nonerythroid cells in peripheral blood or bone marrow are myeloblasts; monocytic precursors are < 20% in bone marrow and granulocytes are 10%+ of cells (Mihova, 2013).
Treatments
Generally, acute myeloid leukemia is treated using chemotherapy consisting of an induction phase and consolidation phase (Dohner et al., 2009). Patients may also consider hematopoietic stem cell transplantation as a second mode of tackling the cancer. The most novel research is being done in tyrosine kinase inhibitors; however M2 acute myeloid leukemia treatment research involves molecules that inhibit the fusion oncoprotein AML1-ETO. Therefore, in terms of M2 subtype acute myeloid leukemia, the most prominent target is the abnormal AML1-ETO fusion protein. Similarly, chronic myeloid leukemia (CML) is comparable to acute myeloid leukemia M2 because it also forms a fusion oncoprotein – BCR-Abl. The developed tyrosine kinase inhibitor, imatinib mesylate, has had a tremendous effect on stopping cancer progression in the majority of chronic myeloid leukemia patients. BCR-Abl is constitutively active due chromosome translocation; therefore it over-phosphorylates the tyrosine kinase. Imatinib mesylate works to block BCR-Abl's activity by blocking the active kinase domain (Fava et al., 2011).
Celastrol is a compound extracted from Tripterygium wilfordii that has anti-cancer properties. It was found to inhibit cell proliferation through the down regulation of AML1-ETO fusion oncoprotein. Celastrol inhibits the fusion oncoprotein by inducing mitochondrial instability and initiating caspase activity. The decrease of AML1-ETO also results in lower levels of C-KIT kinases, Akt/PKB, STAT3, and Erk1/2 – all of which are involved in cell signaling and gene transcription.[3]
Histone deacetylase inhibitors such as valproic acid (VPA), vorinostat, and all-trans retinoic acid (ATRA) are effective in targeting acute myeloid leukemia with the AML1-ETO fusion protein. The HDAC inhibitors are known to induce apoptosis through accumulation of DNA damage, inhibition of DNA repair, and activation of caspases. These inhibitors are extra sensitive to the fusion proteins. Vorinostat has been proven to cause a greater accumulation of DNA damage in fusion protein expressing cells and is directly correlated with the reduction of DNA repair enzymes (Garcia et al., 2008). Romidepsin, a drug in phase two clinical trials, has demonstrated higher efficacy in patients with AML1-ETO fusion protein leukemia (Odenike et al., 2008). Although many clinical evaluations have proven HDAC inhibitors have a promising effect on M2 subtype acute myeloid leukemia, it has not been approved as an official treatment.
In t(6;9) acute myeloid leukemia, FLT3-ITD and the DEK-NUP214 protein are potential targets for treatment. Sorafenib is a kinase inhibitor used as a treatment for kidney and liver cancer. The kinase inhibitor blocks serine-threonine kinase RAF-1 as well as FLT-ITD (Kindler, 2010). The drug has been proven to be effective in reducing FLT3-ITD overexpression (Metzelder et al., 2009). In patients with DEK-NUP214, it was found that the fusion oncoprotein caused an upregulation of mTORC1 (Sanden et al., 2013). Thus, a mTORC inhibitor could be a potential treatment.
References
- "Acute Myeloid Leukemia - Signs and Symptoms".
- Taj AS, Ross FM, Vickers M, et al. (1995). "t(8;21) myelodysplasia, an early presentation of M2 AML". Br. J. Haematol. 89 (4): 890–2. doi:10.1111/j.1365-2141.1995.tb08429.x. PMID 7772527. S2CID 35020709.
- Yu, Xianjun; Ruan, Xuzhi; Zhang, Jingxuan; Zhao, Qun (30 April 2016). "Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia". Molecules. 21 (5): 574. doi:10.3390/molecules21050574. PMC 6274014. PMID 27144550.
- American Cancer Society (ACS) (2016). Cancer Facts and Figures.
- Andrieu V, Radford-Weiss I, Troussard X, Chane C, Valensi F, Guesnu M, et al. (1996). "Molecular detection of t(8;21)/AML1-ETO in AML M1/M2: correlation with cytogenetics, morphology and immunophenotype". British Journal of Haematology. 92 (4): 855–865. doi:10.1046/j.1365-2141.1996.415954.x. PMID 8616078. S2CID 40227895.
- Bennett, J.H. (1852). Leucocythemia or White Cell Blood, pp 7–82. Edinburgh.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C (1976). "Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group". Br J Haematol. 33 (4): 451–8. doi:10.1111/j.1365-2141.1976.tb03563.x. PMID 188440. S2CID 9985915.
- Chi Y, Lindgren V, Quigley S, Gaitonde S (2008). "Acute Myelogenous Leukemia With t(6;9)(p23;q34) and Marrow Basophilia: An Overview". Arch Pathol Lab Med. 132 (11): 1835–7. doi:10.5858/132.11.1835. PMID 18976025.
- Cullen P (1811). "Case of splenitis acutus in which the serum of the blood drawn from the arm had the appearance of milk". Edinburgh Medical Journal: 169–171.
- Dohner H, Estey E, Amadori S, Appelbaum F, Buchner T, Burnett A, et al. (2009). "Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet". Blood. 115 (3): 453–474. doi:10.1182/blood-2009-07-235358. PMID 19880497. S2CID 1088077.
- Elagib K, Goldfarb A (2006). "Oncogenic pathways of AML1-ETO in acute myeloid leukemia: multifaceted manipulation of marrow maturation". Cancer Lett. 251 (2): 179–186. doi:10.1016/j.canlet.2006.10.010. PMC 1931834. PMID 17125917.
- Faderl S, Kantarjian HM, Estey E, Manshouri T, Chan CY, Rahman Elsaied A, et al. (2000). "The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia". Cancer. 89 (9): 1976–1982. doi:10.1002/1097-0142(20001101)89:9<1976::aid-cncr14>3.3.co;2-e. PMID 11064355.
- Falini B, Tiacci E, Martelli MP, Ascani S, Pileri SA (2010). "New classification of acute myeloid leukemia and precursor-related neoplasms: changes and unsolved issues". Discov Med. 10 (53): 281–92. PMID 21034669.
- Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M, Fava C, Nagler A, Della Casa CM, Morra E, Abruzzese E, D'Emilio A, Stagno F, Coutre P, Hurtado-Monroy R, Santini V, Martino B, Pane F, Piccin A, Giraldo P, Assouline S, Durosinmi MA, Leeksma O, Pogliani EM, Puttini M, Jang E, Reiffers J, Valsecchi MG, Kim DW (2011). "Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib". Journal of the National Cancer Institute. 103 (7): 553–561. doi:10.1093/jnci/djr060. PMID 21422402.
- Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. (2008). "Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes". Blood. 111 (3): 1060–6. doi:10.1182/blood-2007-06-098061. PMID 17962510. S2CID 15029739.
- Gilliland D, Griffin J (2000). "The roles of FLT3 in hematopoiesis and leukemia". Blood. 100 (5): 1532–42. doi:10.1182/blood-2002-02-0492. PMID 12176867. S2CID 2362311.
- Francais Groupe, de Cytogenetique Hematoplogique (1990). "Acute myelogenous leukemia with an 8;21 translocation: a report on 148 cases from the Groupe Francais de Cytogenetique Hematologique". Cancer Genetics and Cytogenetics. 44 (2): 169–179. doi:10.1016/0165-4608(90)90043-A.
- Huret, J. t(6;9)(p23;q34). (2005). Atlas Genet Cytogenet Oncol Haematol.
- Kindler T, Lipka DB, Fischer T (2010). "FLT3 as a therapeutic target in AML: still challenging after all these years". Blood. 116 (24): 5089–5102. doi:10.1182/blood-2010-04-261867. PMID 20705759. S2CID 18080783.
- Kobayashi S (2015). "Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy". Biological & Pharmaceutical Bulletin. 38 (8): 1098–103. doi:10.1248/bpb.b15-00096. PMID 26235572.
- Koleva RI, Ficarro SB, Radomska HS, Carrasco-Alfonso MJ, Alberta JA, Webber JT, Luckey CJ, Marcucci G, Tenen DG, Marto JA (2012). "C/EBPa and DEF coordinately regulation myeloid differentiation". Blood. 119 (21): 4878–88. doi:10.1182/blood-2011-10-383083. PMC 3367892. PMID 22474248.
- Köser J, Maco B, Aebi U, Fahrenkrog B (2011). "The nuclear pore complex becomes alive: new insights into its dynamics and involvement in different cellular processes". Atlas of Genetics and Cytogenetics in Oncology and Haematology (2). doi:10.4267/2042/38203.
- Kottaridis P (2001). "The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials" (PDF). Blood. 98 (6): 1752–1759. doi:10.1182/blood.v98.6.1752. PMID 11535508.
- Levy JM, Thorburn A (2011). "Targeting autophagy during cancer therapy to improve clinical outcomes". Pharmacol Ther. 131 (1): 130–141. doi:10.1016/j.pharmthera.2011.03.009. PMC 3539744. PMID 21440002.
- Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, et al. (2009). "Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation". Blood. 113 (26): 6567–6571. doi:10.1182/blood-2009-03-208298. PMID 19389879. S2CID 206878993.
- Mihova, D. (2013). Leukemia - Acute - Acute myeloid leukemia with maturation (FAB AML M2). Pathologyoutlines.com. Retrieved 5 May 2016, from http://www.pathologyoutlines.com/topic/leukemiaM2.html
- Miyoshi H.; et al. (1991). "t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1". Proc. Natl. Acad. Sci. USA. 88 (23): 10431–10434. Bibcode:1991PNAS...8810431M. doi:10.1073/pnas.88.23.10431. PMC 52942. PMID 1720541.
- O'Donnell M, Abboud C, Altman J, Appelbaum F, Arber D, Attar E, et al. (2012). "Acute Myeloid Leukemia". Journal of the National Comprehensive Cancer Network. 10 (8): 984–1021. doi:10.6004/jnccn.2012.0103. PMID 22878824.
- Odenike OM, Alkan S, Sher D, Godwin JE, Huo D, Brandt SJ, et al. (2008). "Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia". Clinical Cancer Research. 14 (21): 7095–101. doi:10.1158/1078-0432.ccr-08-1007. PMC 4498482. PMID 18981008.
- Sandén C, Ageberg M, Petersson J, Lennartsson A, Gullberg U (2013). "Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR". BMC Cancer. 13 (1): 440. doi:10.1186/1471-2407-13-440. PMC 3849736. PMID 24073922.
- Schwartz S, Jiji R, Kerman S, Meekins J, Cohen MM (1983). "Translocation (6;9)(p23; q34) in acute nonlymphocytic leukemia". Cancer Genet Cytogenet. 10 (2): 133–138. doi:10.1016/0165-4608(83)90116-4. PMID 6616433.
- Song G, Ouyang G, Bao S (2005). "The activation of Akt/PKB signalling pathway and cell survival". J Cell Mol Med. 9 (1): 59–71. doi:10.1111/j.1582-4934.2005.tb00337.x. PMC 6741304. PMID 15784165.
- Torgersen M, Engedal N, Boe S, Hokland P, Simonsen A (2013). "Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells". Blood. 122 (14): 2467–2476. doi:10.1182/blood-2013-05-500629. PMID 23970379. S2CID 25009615.
- Velpeau A (1827). "Sur la resorptiendu pus et sur l'alteration du sang dans les malades". Revue Medicine. 2: 216–218.
- Virchow R (1845). "Weisses Blut". Froriep's Notizen. 36: 151–156.
- Weber J, Taylor L, Roussel M, Sherr C, Bar-Sagi D (1999). "Nucleolar Arf sequesters Mdm2 and activates p53". Nature Cell Biology. 1 (1): 20–26. doi:10.1038/8991. PMID 10559859. S2CID 25132981.
- Weinberg, R. (2014). The Biology of Cancer (2nd ed.). New York: Garland Science.
- Yu X, Ruan X, Zhang J, Zhao Q (2016). "Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia". Molecules. 21 (5): 574. doi:10.3390/molecules21050574. PMC 6274014. PMID 27144550.