Migalastat
Migalastat, sold under the brand name Galafold, is a medication for the treatment of Fabry disease, a rare genetic disorder. It was developed by Amicus Therapeutics. The US Food and Drug Administration (FDA) granted it orphan drug status in 2004,[3] and the European Commission followed in 2006.[4] The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) granted the drug a marketing approval under the name Galafold in May 2016.[5][6][7]
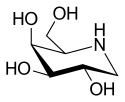 | |
| Clinical data | |
|---|---|
| Pronunciation | mi GAL a stat |
| Trade names | Galafold |
| Other names | DDIG, AT1001, 1-deoxygalactonojirimycin |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 75% |
| Protein binding | None |
| Metabolites | O-glucuronides (<15%) |
| Elimination half-life | 3–5 hours (single dose) |
| Excretion | Urine (77%), feces (20%) |
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C6H13NO4 |
| Molar mass | 163.173 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
Medical uses
Migalastat is used for the long-term treatment of Fabry disease in adults and adolescents aged 16 or older with an amenable mutation of the enzyme alpha-galactosidase A (α-GalA). An "amenable" mutation is one that leads to misfolding of the enzyme, but otherwise would not significantly impair its function.[6]
Based on an in vitro test, Amicus Therapeutics has published a list of 269 amenable and nearly 600 non-amenable mutations. About 35 to 50% of people with Fabry have an amenable mutation.[6]
Adverse effects
The most common side effect in clinical trials was headache (in about 10% of people who take it). Less common side effects (between 1 and 10% of people) included unspecific symptoms such as dizziness, fatigue, and nausea, but also depression. Possible rare side effects could not be assessed because of the low number of subjects in the clinical trials in which adverse effects were measured.[6]
Interactions
When combined with intravenous agalsidase alfa or beta, which are recombinant versions of the enzyme α-GalA, migalastat increases tissue concentrations of functional α-GalA compared to agalsidase given alone up to fivefold.Migalastat is not intended to be combined with agalsidase.[6]
Migalastat does not inhibit or induce cytochrome P450 liver enzymes or transporter proteins and is therefore expected to have a low potential for interactions with other drugs.[6]
Pharmacology
Mechanism of action
Fabry disease is a genetic disorder caused by various mutations of the enzyme α-GalA, which is responsible for breaking down the sphingolipid globotriaosylceramide (Gb3), among other glycolipids and glycoproteins. Some of these mutations result in misfolding of α-GalA, which subsequently fails protein quality control in the endoplasmic reticulum and is decomposed. Lack of functional α-GalA leads to accumulation of Gb3 in blood vessels and other tissues, with a wide range of symptoms including kidney, heart, and skin problems.[9]
Migalastat is a potent, orally available inhibitor of α-GalA (IC50: 4 μM).[9] When binding to faulty α-GalA, it shifts the folding behaviour towards the proper conformation, resulting in a functional enzyme provided the mutation is amenable.[6] Molecules with this type of mechanism are called pharmacological chaperones.[9]
When the enzyme reaches its destination, the lysosome, migalastat dissociates because of the low pH and the relative abundance of Gb3 and other substrates, leaving α-GalA free to fulfill its function. Depending on the mutation, the EC50 is between 0.8 μM and over 1 mM in cellular models.[10]
 The enzyme alpha-galactosidase A (α-GalA)
The enzyme alpha-galactosidase A (α-GalA) Globotriaosylceramide (Gb3), a substrate of α-GalA, has a terminal D-galactose structurally similar to migalastat.[11]
Globotriaosylceramide (Gb3), a substrate of α-GalA, has a terminal D-galactose structurally similar to migalastat.[11] Migalastat ("top" view)
Migalastat ("top" view)
Pharmacokinetics
Migalastat is almost completely absorbed from the gut; taking the drug together with food decreases its absorption by about 40%. Total bioavailability is about 75% when taken without food. The substance is not bound to blood plasma proteins.[6]
Only a small fraction of a migalastat dose is metabolized, mainly to three dehydrogenated O-glucuronides (4% of the dose) and a number of unspecified metabolites (10%). The drug is mainly eliminated via the urine (77%) and to a smaller extent via the faeces (20%). Practically all of the metabolites are excreted in the urine. Elimination half-life is three to five hours after a single dose.[6]
Chemistry
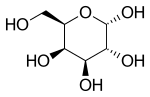
Migalastat is used in form of the hydrochloride, which is a white crystalline solid and is soluble in water.[12]: 11 The molecule has four asymmetric carbon atoms with the same stereochemistry as the sugar D-galactose, but is missing the first hydroxyl group. It has a nitrogen atom in the ring instead of an oxygen, which makes it an iminosugar.[13]
The structure is formally derived from nojirimycin.
History
Migalastat was isolated as a fermentation product of the bacterium Streptomyces lydicus (strain PA-5726) in 1988 and called 1-deoxygalactonojirimycin.[13][14] In 2004, it was designated orphan drug status by the US FDA for the treatment of Fabry disease,[3] and in 2006 the European CHMP did likewise.[15] The sponsorship for the drug was transferred several times over the following years: from Amicus Therapeutics to Shire Pharmaceuticals in 2008, back to Amicus in 2010, to Glaxo in 2011, and again to Amicus in 2014.[16]
Two phase III clinical trials with a total of about 110 subjects were conducted between 2009 and 2015, one double-blind comparing the drug to placebo, and one comparing it to recombinant α-GalA without blinding. Migalastat stabilised heart and kidney function over the 30-months period of these trials.[6]
In September 2015, Amicus announced that it would submit a new drug application (NDA) for accelerated approval of migalastat to the FDA by the end of 2015.[17] The CHMP recommended approval in April 2016, but the FDA rejected the application in November for having insufficient data in November 2016.[18] The drug was approved in the European Union in May 2016.[5] Germany was the first country where migalastat was launched.[5] After Scott Gottlieb became FDA commissioner in 2017, the CEO of Amicus began lobbying him directly for the FDA to accept the NDA and in February 2018 the FDA accepted it and promised a response by August 2018.[19]
See also
- Miglustat, a drug for the treatment of Gaucher disease, with a similar structure
- 1-Deoxynojirimycin, a stereoisomer of migalastat
References
- "Galafold (Amicus Therapeutics Pty Ltd)". tga.gov.au. Retrieved 29 March 2023.
- "Prescription medicines: registration of new chemical entities in Australia, 2017". Therapeutic Goods Administration (TGA). 21 June 2022. Retrieved 9 April 2023.
- "Migalastat Orphan Drug Designations and Approvals". U.S. Food and Drug Administration (FDA). Retrieved 16 September 2020.
- "EU/3/06/368". European Medicines Agency (EMA). 17 September 2018. Retrieved 16 September 2020.
- "Amicus Therapeutics Announces European Commission Approval for Galafold (Migalastat) in Patients with Fabry Disease in European Union". GlobeNewswire. 31 May 2016.
- "Summary of Product Characteristics for Galafold" (PDF). European Medicines Agency. June 2016.
- "Galafold EPAR". European Medicines Agency (EMA). 17 September 2018. Retrieved 16 September 2020.
- New Drug Therapy Approvals 2018 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2019. Retrieved 16 September 2020.
- Sánchez-Fernández EM, García Fernández JM, Mellet CO (April 2016). "Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: lessons from Gaucher, GM1-gangliosidosis and Fabry diseases" (PDF). Chemical Communications. 52 (32): 5497–515. doi:10.1039/C6CC01564F. PMID 27043200.
- Benjamin ER, Flanagan JJ, Schilling A, Chang HH, Agarwal L, Katz E, et al. (June 2009). "The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines". Journal of Inherited Metabolic Disease. 32 (3): 424–40. doi:10.1007/s10545-009-1077-0. PMID 19387866. S2CID 12629461.
- Warnock DG, Bichet DG, Holida M, Goker-Alpan O, Nicholls K, Thomas M, et al. (2015). "Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients when Co-Administered with Infused Agalsidase". PLOS ONE. 10 (8): e0134341. Bibcode:2015PLoSO..1034341W. doi:10.1371/journal.pone.0134341. PMC 4529213. PMID 26252393.
- "Assessment report EMA/272226/2016" (PDF). EMA. 1 April 2016.
- Asano, N (2007). "Naturally occurring iminosugars and related alkaloids: structure, activity and applications". In Compain, P; Martin, OR (eds.). Iminosugars: from synthesis to therapeutic applications. Wiley and Sons. p. 17. ISBN 978-0-470-03391-3.
- Miyake Y, Ebata M (1988). "The structures of a β-galactosidase inhibitor, Galactostatin, and its derivatives". Agric Biol Chem. 52 (3): 661–666. doi:10.1271/bbb1961.52.661.
- "Galafold". European Medicines Agency. 1 April 2016.
- "Public summary of opinion on orphan designation". European Medicines Agency. 29 April 2014.
- "Amicus Therapeutics Plans to Submit NDA for Migalastat for Fabry Disease Following Positive Pre-NDA Meeting With FDA". Drugs.com. 15 September 2015.
- Adams B (November 29, 2016). "FDA rejects quick Amicus Fabry drug read-through, new data not expected till 2019". FierceBiotech.
- Carroll J (February 12, 2018). "Once rejected, FDA now rolls out a short red carpet for Amicus' migalastat". Endpoints.
External links
- "Migalastat". Drug Information Portal. U.S. National Library of Medicine.