Multiscale Green's function
Multiscale Green's function (MSGF) is a generalized and extended version of the classical Green's function (GF) technique[1] for solving mathematical equations. The main application of the MSGF technique is in modeling of nanomaterials.[2] These materials are very small – of the size of few nanometers. Mathematical modeling of nanomaterials requires special techniques and is now recognized to be an independent branch of science.[3] A mathematical model is needed to calculate the displacements of atoms in a crystal in response to an applied static or time dependent force in order to study the mechanical and physical properties of nanomaterials. One specific requirement of a model for nanomaterials is that the model needs to be multiscale and provide seamless linking of different length scales.[4]
Green's function (GF) was originally formulated by the British mathematical physicist George Green in the year 1828 as a general technique for solution of operator equations.[1] It has been extensively used in mathematical Physics over the last almost two hundred years and applied to a variety of fields.[1][5] Reviews of some applications of GFs such as for many body theory and Laplace equation are available in the Wikipedia. The GF based techniques are used for modeling of various physical processes in materials such as phonons,[6] Electronic band structure[7] and elastostatics.[5]
Application of the MSGF method for modeling nanomaterials
The MSGF method is a relatively new GF technique for mathematical modeling of nanomaterials. Mathematical models are used for calculating the response of materials to an applied force in order to simulate their mechanical properties. The MSGF technique links different length scales in modeling of nanomaterials.[2][8] Nanomaterials are of atomistic dimensions and need to be modeled at the length scales of nanometers. For example, a silicon nanowire, whose width is about five nanometers, contains just 10 – 12 atoms across its width. Another example is graphene[9] and many new two-dimensional (2D) solids.[10] These new materials are ultimate in thinness because they are just one or two atoms thick. Multiscale modeling is needed for such materials because their properties are determined by the discreteness of their atomistic arrangements as well as their overall dimensions.[2][4]
The MSGF method is multiscale in the sense that it links the response of materials to an applied force at atomistic scales to their response at the macroscopic scales. The response of materials at the macroscopic scales is calculated by using the continuum model of solids. In the continuum model, the discrete atomistic structure of solids is averaged out into a continuum. Properties of nanomaterials are sensitive to their atomistic structure as well as their overall dimensions. They are also sensitive to the macroscopic structure of the host material in which they are embedded. The MSGF method is used to model such composite systems.
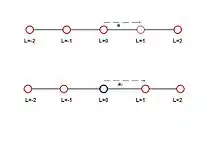
The MSGF method is also used for analyzing behavior of crystals containing lattice defects such as vacancies, interstitials, or foreign atoms. Study of these lattice defects is of interest as they play a role in materials technology.[11][12] Presence of a defect in a lattice displaces the host atoms from their original position or the lattice gets distorted. This is shown in Fig 1 for a 1D lattice as an example. Atomistic scale modeling is needed to calculate this distortion near the defect,[13][14] whereas the continuum model is used to calculate the distortion far away from the defect. The MSGF links these two scales seamlessly.
MSGF for nanomaterials
The MSGF model of nanomaterials accounts for multiparticles as well as multiscales in materials.[8] It is an extension of the lattice statics Green’s function (LSGF) method that was originally formulated at the Atomic Energy Research Establishment Harwell in U.K. in 1973.[11][15] It is also referred to as Tewary method in the literature[16][17] The LSGF method complements molecular dynamics[18] (MD) method for modeling multiparticle systems. The LSGF method is based upon the use of the Born von Karman (BvK) model[6][19] and can be applied to different lattice structures and defects.[11][17][20] The MSGF method is an extended version of the LSGF method and has been applied to many nanomaterials and 2D materials[2]
At the atomistic scales, a crystal or a crystalline solid is represented by a collection of interacting atoms located at discrete sites on a geometric lattice.[19] A perfect crystal consists of a regular and periodic geometric lattice. The perfect lattice has translation symmetry, which means that all the unit cells are identical. In a perfect periodic lattice, which is assumed to be infinite, all atoms are identical. At equilibrium each atom is assumed to be located at its lattice site. The force at any atom due to other atoms just cancels out so the net force at each atom is zero. These conditions break down in a distorted lattice in which atoms get displaced from their positions of equilibrium.[15] The lattice distortion may be caused by an externally applied force. The lattice can also be distorted by introducing a defect in the lattice or displacing an atom that disturbs the equilibrium configuration and induces a force on the lattice sites. This is shown in Fig. 1. The objective of the mathematical model is to calculate the resulting values of the atomic displacements.
The GF in the MSGF method is calculated by minimizing the total energy of the lattice.[15] The potential energy of the lattice in the form of an infinite Taylor series in atomic displacements in the harmonic approximation as follows

where L and L′ label the atoms, a and b denote the Cartesian coordinates, u denotes the atomic displacement, and −f and K are the first and second coefficients in the Taylor series. They are defined by[1]

and

where the derivatives are evaluated at zero displacements. The negative sign is introduced in the definition of f for convenience. Thus f(L) is a 3D vector that denotes the force at the atom L. Its three Cartesian components are denoted by fa(L) where a = x, y, or z. Similarly K(L,L’) is a 3x3 matrix, which is called the force- constant matrix between the atoms at L and L’. Its 9 elements are denoted by Kab(L,L′) for a, b = x, y, or z.
At equilibrium, the energy W is minimum.[8] Accordingly, the first derivative of W with respect to each u must be zero. This gives the following relation from Eq. (1)

It can be shown by direct substitution that the solution of Eq. (4) can be written as

where G is defined by the following inversion relation

In Eq. (6), δ(m,n) is the discrete delta function of two discrete variable m and n. Similar to the case of Dirac delta function for continuous variables, it is defined to be 1 if m = n and 0 otherwise.[6]
Equations (4)–(6) can be written in the matrix notation as follows:

The matrices K and G in the above equations are 3N × 3N square matrices and u and f are 3N-dimensional column vectors, where N is the total number of atoms in the lattice. The matrix G is the multiparticle GF and is referred to as the lattice statics Green’s function (LSGF),.[15] If G is known, the atomic displacements for all atoms can be calculated by using Eq. (8).
One of the main objectives of modeling is the calculation of the atomistic displacements u caused by an applied force f. [21] The displacements, in principle, are given by Eq. (8). However, it involves inversion of the matrix K which is 3N x 3N. For any calculation of practical interest N ~ 10,000 but preferably a million for more realistic simulations. Inversion of such a large matrix is computationally extensive and special techniques are needed for the calculation of u’s. For regular periodic lattices, LSGF is one such technique. It consists of calculating G in terms of its Fourier transform, and is similar to the calculation of the phonon GF.[6]
The LSGF method has now been generalized to include the multiscale effects in the MSGF method.[8] The MSGF method is capable of linking length scales seamlessly. This property has been used in developing a hybrid MSGF method that combines the GF and the MD methods and has been used for simulating less symmetric nanoinclusions such as quantum dots in semiconductors.[22]
For a perfect lattice without defects, the MSGF links directly the atomistic scales in LSGF to the macroscopic scales through the continuum model. A perfect lattice has full translation symmetry so all the atoms are equivalent. In this case any atom can be chosen as the origin and G(L,L') can be expressed by a single index (L'-L)[6] defined as

The asymptotic limit of G(L), that satisfies Eq. (10), for large values of R(L) is given by[8]
![{\displaystyle \lim _{R(L)\to \infty }[G(0,L)]\rightarrow G_{c}(x)+O(1/x^{4})\qquad \qquad (11)}](../I/6649f3f37f35655d6f705dbfe1979c0ec4c03e6a.svg)
where x = R(L) is the position vector of the atom L, and Gc(x) is the continuum Green's function (CGF), which is defined in terms of the elastic constants and used in modeling of conventional bulk materials at macroscales.[5][11] In Eq. (11), O(1/xn) is the standard mathematical notation for a term of order 1/xn and higher. The magnitude of Gc(x) is O(1/x2).[21] The LSGF G(0,L) in this equation reduces smoothly and automatically to the CGF for large enough x as terms O(1/x4) become gradually small and negligible. This ensures the seamless linkage of the atomistic length scale to the macroscopic continuum scale.[8]
Equations (8) and (9) along with the limiting relation given by Eq. (11), form the basic equations for the MSGF.[8] Equation (9) gives the LSGF, which is valid at the atomistic scales and Eq. (11) relates it to the CGF, which is valid at the macro continuum scales. This equation also shows that the LSGF reduces seamlessly to the CGF.
MSGF method for calculating the effect of defects and discontinuities in nanomaterials
If a lattice contains defects, its translation symmetry is broken. Consequently, it is not possible to express G in terms of a single distance variable R(L). Hence Eq. (10) is not valid anymore and the correspondence between the LSGF and the CGF, needed for their seamless linking breaks down.[15] In such cases the MSGF links the lattice and the continuum scales by using the following procedure:[15]
If p denotes the change in the matrix K, caused by the defect(s), the force constant matrix K* for the defective lattice is written as

As in the case for the perfect lattice in Eq. (9), the corresponding defect GF is defined as the inverse of the full K* matrix. Use of Eq. (12), then leads to the following Dyson’s equation for the defect LSGF:[15]

The MSGF method consists of solving Eq. (13) for G* by using the matrix partitioning technique or double Fourier transform.[6]
Once G* is known, the displacement vector is given by the following GF equation similar to Eq. (8):
u= G* f (14)
Equation (14) gives the desired solution, that is, the atomic displacements or the lattice distortion caused by the force f. However, it does not show the linkage of the lattice and the continuum multiple scales, because Eqs. (10) and (11) are not valid for the defect LSGF G*. The linkage between the lattice and the continuum model in case of lattice with defects is achieved by using an exact transformation described below.[8]
Using Eq.(13), Eq. (14) can be written in the following exactly equivalent form:
u = Gf + G p G* f . (15)
Use of Eq. (14) again on the right hand side of Eq. (15) gives,
u = G f* (16)
where
f* = f + p u. (17)
Note that Eq. (17) defines an effective force f* such that Eqs. (14) and (16) are exactly equivalent.
Equation (16) expresses the atomic displacements u in terms of G, the perfect LSGF even for lattices with defects. The effect of the defects is included exactly in f*. The LSGF G is independent of f or f* and reduces to the CGF asymptotically and smoothly as given in Eq. (11). The effective force f* can be determined in a separate calculation by using an independent method if needed, and the lattice statics or the continuum model can be used for G. This is the basis of a hybrid model that combines MSGF and MD for simulating a germanium quantum dot in a silicon lattice.[22]
Equation (16) is the master equation of the MSGF method.[2][8] It is truly multiscale. All the discrete atomistic contributions are included in f*. The Green's function G can be calculated independently, which can be fully atomistic for nanomaterials or partly or fully continuum for macroscales to account for surfaces and interfaces in material systems as needed [8]
GF as Physical Characteristic of Solids
Tewary, Quardokus and DelRio[23] have suggested that Green's function is not just a mathematical artefact, but a physical characteristic of the solid, which can be measured by using scanning probe microscopy.
Their suggestion is based upon the fact that any process of measurement on a system, requires quantifying the response of the system to an external probe and the Green’s function gives the total response of the system to an applied probe.[24] For example, if we want to measure the characteristics of a spring, we fix it at one end and apply a force f or f* at the other end. A measurement of the stretching u of the spring will then give the value of the Green’s function of the spring by using Eq. 14 or 16. In this example, the applied force is the probe and the stretching of the spring is its response to the probe. If the measured values of the Green’s functions of a solid are available, it will accurately characterize the response of a solid for engineering applications. For this reason, the Green’s function is also called the response function.
Causal Green's Function Molecular Dynamics
An important application of the MSGF method is in modeling the temporal (time-dependent) processes in solids, especially in nanomaterials. This is needed in diverse applications like testing and characterization of materials, propagation of waves and heat in nanomaterials, and modeling of radiation damage in semiconductors.[2][18] These processes need to be simulated over a wide range of times from femtoseconds to nanoseconds or even microseconds which is a challenging multiscale problem for nanomaterials. It was shown by Tewary[25] that the use of the causal Green’s functions in molecular dynamics can significantly accelerate the temporal convergence of molecular dynamics. The new method, called CGFMD (Causal Green's Function Molecular Dynamics) is the temporal equivalent of the MSGF and is based upon the use of causal or retarded Green's functions. It has been applied[25] to simulate the propagation of ripples in graphene,[9] where it has been shown that the CGFMD can model time scales over 6 to 9 orders of magnitude at the atomistic level. At least in some idealized cases such as propagation of ripples in graphene, the CGFMD can bridge the time scales from femto to microseconds. The CGFMD has been refined and further developed in papers by Coluci, Dantas and Tewary.[26][27]}
See also
References
- Morse, Philip; Feshbach, Herman (1953). Methods of Theoretical Physics. New York: McGraw-Hill Publishing Company.
- Tewary, Vinod; Zhang, Yong (2015). Modeling, Characterization and Production of Nanomaterials. Amsterdam: Elsevier.
- Rapp, Bob (2005). "Third branch of science (Modeling)". Materials Today. 8 (January): 6.
- Karakasidis, T.; Charitidis, C. (2007). "Multiscale modeling in nanomaterials science". Materials Science and Engineering. 27 (5–8): 1082–1089. doi:10.1016/j.msec.2006.06.029.
- Pan, Ernian; Chen, Weiqiu (2015). Static Green's functions in Anisotropic Media. New York: Cambridge University Press.
- Maradudin, A.; Montroll, E.; Weiss, G.; Ipatova, I. (1971). Theory of lattice dynamics in the harmonic approximation. Solid State Physics. Vol. Supplement 3 (Second ed.). New York: Academic Press.
- Callaway, J. (1964). Energy band theory. New York: Academic Press.
- Tewary, Vinod (2004). "Multiscale Green's-function method for modeling point defects and extended defects in anisotropic solids". Physical Review B. 69: 13. doi:10.1103/physrevb.69.094109.
- Fasolino, A.; Los, J.; Katsnelson, M. (2007). "Intrinsic ripples in graphene". Nature Materials. 6 (11): 858–861. arXiv:0704.1793. Bibcode:2007NatMa...6..858F. doi:10.1038/nmat2011. PMID 17891144. S2CID 38264967.
- Mas-Balleste, R.; Gomez-Navarro, C.; Gomez-Herrero, J.; Zamora, F. (2011). "2D materials: to graphene and beyond". Nanoscale. 3 (1): 20–30. Bibcode:2011Nanos...3...20M. doi:10.1039/c0nr00323a. PMID 20844797.
- Stoneham, A. (2001). Theory of defects in solids. Oxford: Clarendon Press.
- Ebert, P. (2002). "Defects in III–V semiconductor surfaces". Applied Physics A: Materials Science & Processing. 75 (1): 101–112. Bibcode:2002ApPhA..75..101E. doi:10.1007/s003390101059. S2CID 43938452.
- Bullough, R.; Hardy, J. (1968). "Strain Field Interaction between Vacancies in Copper and Aluminium". Philosophical Magazine. 17 (148): 833–842. Bibcode:1968PMag...17..833B. doi:10.1080/14786436808223032.
- Kanzaki, H. (1957). "Point defects in face-centered cubic lattice". J. Phys. Chem. Solids. 2: 24–36. doi:10.1016/0022-3697(57)90003-3.
- Tewary, V. (1973). "Green-function method for lattice statics". Advances in Physics. 22 (6): 757–810. Bibcode:1973AdPhy..22..757T. doi:10.1080/00018737300101389.
- Ben-Abraham, S.; Rabinovich, A.; Pelleg, J. (1977). "Relations between Vacancy Migration and Formation Energies, Debye Temperature and Melting Point". Physica Status Solidi B. 84 (2): 435–441. Bibcode:1977PSSBR..84..435B. doi:10.1002/pssb.2220840205.
- Glass, N.; Boffi, S.; Bilellof, J. (1977). "Inelastic neutron scattering from screw dislocations". J. Phys. C: Solid State Phys. 10 (13): 2307–2319. Bibcode:1977JPhC...10.2307G. doi:10.1088/0022-3719/10/13/007.
- Rapaport, D. (2004). The art of molecular dynamics simulation. Cambridge, UK: Cambridge University Press.
- Kittel, C. (1996). Introduction to Solid State Physics. New York: John Wiley.
- Thomson, R.; Zhou, S.; Carlsson, A. (1992). "Lattice Imperfections Studied by Use of Lattice Green-Functions". Physical Review B. 46 (17): 10613–10622. Bibcode:1992PhRvB..4610613T. doi:10.1103/physrevb.46.10613. PMID 10002913.
- Eshelby, J. (1956). "The Continuum Theory of Lattice Defects". Solid State Physics. 3: 79–114. doi:10.1016/S0081-1947(08)60132-0. ISBN 9780126077032.
- Read, D. (2007). "Multiscale model of near-spherical germanium quantum dots in silicon". Nanotechnology. 18 (10): 105402. Bibcode:2007Nanot..18j5402R. doi:10.1088/0957-4484/18/10/105402. S2CID 55125668.
- Tewary, V. K.; Quardokus, Rebecca C.; DelRio, Frank W. (29 April 2016). "Green's function modeling of response of two-dimensional materials to point probes for scanning probe microscopy". Physics Letters A. 380 (20): 1750–1756. Bibcode:2016PhLA..380.1750T. doi:10.1016/j.physleta.2016.03.021. PMC 5520644. PMID 28736478.
- Tewary, Vinod; Zhang, Yong (2015). Modeling, characterization and production of nanomaterials : electronics, photonics and energy applications. Cambridge, UK. ISBN 978-1-78242-228-0.
{{cite book}}: CS1 maint: location missing publisher (link) - Tewary, V. K. (22 October 2009). "Extending the time scale in molecular dynamics simulations: Propagation of ripples in graphene". Physical Review B. 80 (16): 161409. Bibcode:2009PhRvB..80p1409T. doi:10.1103/PhysRevB.80.161409.
- Coluci, V. R.; Dantas, S. O.; Tewary, V. K. (29 May 2018). "Generalized Green's function molecular dynamics for canonical ensemble simulations". Physical Review E. 97 (5): 053310. Bibcode:2018PhRvE..97e3310C. doi:10.1103/PhysRevE.97.053310. PMID 29906874. S2CID 49219692.
- Coluci, V.R.; Dantas, S.O.; Tewary, V.K. (August 2022). "Accelerated causal Green's function molecular dynamics". Computer Physics Communications. 277: 108378. Bibcode:2022CoPhC.27708378C. doi:10.1016/j.cpc.2022.108378. S2CID 248348337.