N-sulfoglucosamine sulfohydrolase
In enzymology, a N-sulfoglucosamine sulfohydrolase (EC 3.10.1.1), otherwise known as SGSH, is an enzyme that catalyzes the chemical reaction
- N-sulfo-D-glucosamine + H2O D-glucosamine + sulfate

| N-sulfoglucosamine sulfohydrolase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
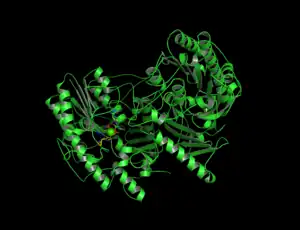 Cartoon depiction of N-sulfoglucosamine sulfohydrolase. | |||||||||
| Identifiers | |||||||||
| EC no. | 3.10.1.1 | ||||||||
| CAS no. | 37289-41-1 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Thus, the two substrates of this enzyme are N-sulfo-D-glucosamine and H2O, whereas its two products are D-glucosamine and sulfate.
This enzyme belongs to the family of hydrolases, specifically those acting on sulfur-nitrogen bonds. The systematic name of this enzyme class is N-sulfo-D-glucosamine sulfohydrolase. Other names in common use include sulfoglucosamine sulfamidase, heparin sulfamidase, 2-desoxy-D-glucoside-2-sulphamate sulphohydrolase (sulphamate, and sulphohydrolase). This enzyme participates in glycosaminoglycan degradation and glycan structures - degradation.
Structure
N-glusoamine sulfohydrolase is a homodimer of two identical monomeric subunits that associate non-covalently. The crystal structure, solved using molecular replacement, consists of two domains: a large N-terminal domain (domain 1) and a smaller C-terminal domain (domain 2) that are both centered about a β-sheet. The core of Domain 1 is formed by eight β-strands surrounded by nine ⍺-helices while the core of Domain 2 is formed by a four-stranded antiparallel β-sheet surrounded by 4 ⍺-helices, accompanied by a C-terminal extension of a small two-stranded β-sheet. There are two disulfide bonds that serve to stabilize a large loop segment in Domain 1 and connect the C-terminal extension to a nearby loop in Domain 2.
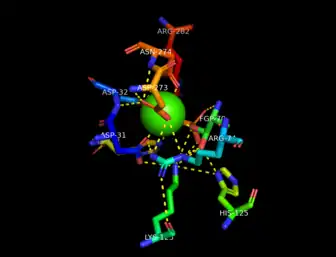
The active site is located through a short tunnel at the bottom of the surface cleft of the enzyme close to the end of the first β-strand in Domain 1 of the dimer subunits. A calcium ion, Ca2+, is coordinated in an octahedral arrangement by oxygen atoms of nearby side chains of residues Asp31, Asp32, Asp273, Asn274, and phosphorylated formylglycine, PFGly70. The PFGly70, which is post-translationally converted from a cysteine and is key in the catalytic activity of the enzyme, is stabilized by the side chains of residues Arg74, Lys123, His125, His181, Asp273, and Arg282 and the coordinate calcium ion.[1][2]
Mechanism
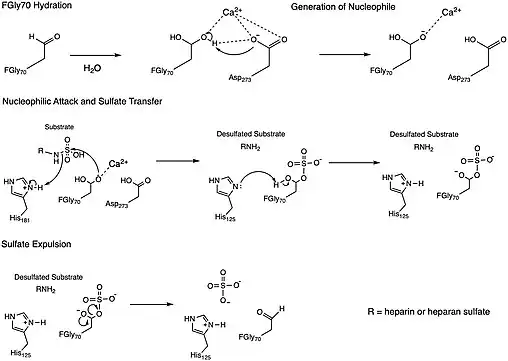
One of the proposed mechanisms for the N-sulfoglucosamine sulfohydrolase, depicted in the figure above, was inspired by a mechanism determined for a sulfatase enzymes, which similarly cleave sulfate ester groups from their substrates.[4] The N-sulfoglucosamine sulfohydrolase mechanism involves four key residues found in the enzyme active site: formylglycine (FGly70), two histidines (His125 and His181), and aspartic acid (Asp273). The formylglycine residue (FGly70) is first hydrated to form a geminal diol whose hydroxyl group subsequently coordinates with a Calcium(II) ion in the active site. A neighboring aspartic acid residue (Asp273) then acts as a base to deprotonate the coordinated hydroxyl group in the geminal diol, which acts as a nucleophile to the sulfate group on the substrate and leads to the cleavage of the substrate nitrogen-sulfur bond. A proximal histidine residue (His181) is proposed to donate a proton as a replacement for the sulfate group that was involved in the nitrogen-sulfur bond. At this point, the formylglycine (FGly70) has the sulfate group attached adjacent to a hydroxyl group, which is believed to be deprotonated by a second histidine group (His125) so that the negatively-charged oxygen atom can participate in the removal of the sulfate group from the residue. This final step liberates the enzyme from the sulfate group.[2]
This mechanism, however, is still being studied for N-sulfoglucosamine sulfohydrolase, meaning other mechanisms are still plausible for the reaction.[2] For instance, a second mechanism proposal involves the aldehyde form of the formylglycine residue (FGly70) serving as an electrophile as it is attacked by one of the oxygen atoms on the sulfate group to transfer the sulfate from the substrate to the enzyme.[4] The order of attack for the sulfate transfer step in this proposed mechanism is therefore inverted to that of the proposed mechanism above.
Overall, the N-sulfoglucosamine sulfohydrolase mechanisms proposed involve the cleavage of the substrate nitrogen-sulfur bond - transferring the sulfate group from the substrate to the enzyme - and freeing the enzyme from its bond to the transferred sulfate group.[4][2]
Function
As previously noted, N-sulfoglucosamine sulfohydrolase plays a crucial role in the degradation of glycosaminoglycans (GAGs), including heparin and heparan sulfate.[5][1] Since these GAGs, particularly haparan sulfates, are integral to several biochemical processes, such as signaling pathways, any disruption in N-sulfoglucosamine sulfohydrolase activity could lead to serious diseases as noted below.[5]
Disease relevance
Sanfillipo Syndrome or Mucopolysaccharidosis III, MPS III, is a lysosomal storage disease resulting from a deficiency in one of five lysosomal enzymes: N-glusoamine sulfohydrolase (Type A), a-N-acetylglucoaminidase (Type B), acetyl CoA a-glusoaminide acetyltransferase (Type C), a-N-acetylglusoamine 6-sulfatase (Type D), and N-glusoamine 3-O-sulfatase (Type E) caused by a dysfunction of one of the genes encoding the enzyme.[6][5][7] These enzymes are responsible for the degradation of heparin sulfate which, in MPS III, accumulates in the lysosomes and outside of the cell, as the primary storage material.[8] In Mucopolysaccharidosis type IIIA, where there are genetic changes in the SGSH gene, there are initial signs of neurodegeneration, developmental delays, and behavioral problems with a wide phenotypic variability.[9] This is the most common form of Mucopolysaccharidosis III with a prevalence of 1 in every 100,000 individuals.[10]
References
- Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K (April 1999). "Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases". The EMBO Journal. 18 (8): 2084–2091. doi:10.1093/emboj/18.8.2084. PMC 1171293. PMID 10205163.
- Sidhu NS, Schreiber K, Pröpper K, Becker S, Usón I, Sheldrick GM, et al. (May 2014). "Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA". Acta Crystallographica. Section D, Biological Crystallography. 70 (Pt 5): 1321–1335. doi:10.1107/S1399004714002739. PMC 4014121. PMID 24816101.
- Madej T, Lanczycki CJ, Zhang D, Thiessen PA, Geer RC, Marchler-Bauer A, Bryant SH (January 2014). "MMDB and VAST+: tracking structural similarities between macromolecular complexes". Nucleic Acids Research. 42 (Database issue): D297–D303. doi:10.1093/nar/gkt1208. PMC 3965051. PMID 24319143.
- Boltes I, Czapinska H, Kahnert A, von Bülow R, Dierks T, Schmidt B, et al. (June 2001). "1.3 A structure of arylsulfatase from Pseudomonas aeruginosa establishes the catalytic mechanism of sulfate ester cleavage in the sulfatase family". Structure. 9 (6): 483–491. doi:10.1016/S0969-2126(01)00609-8. PMID 11435113.
- Webber DL, Choo A, Hewson LJ, Trim PJ, Snel MF, Hopwood JJ, et al. (May 2018). "Neuronal-specific impairment of heparan sulfate degradation in Drosophila reveals pathogenic mechanisms for Mucopolysaccharidosis type IIIA". Experimental Neurology. 303: 38–47. doi:10.1016/j.expneurol.2018.01.020. PMID 29408731. S2CID 3545023.
- Gorbunova VN, Buchinskaia NV (2021-12-13). "Lysosomal storage diseases. Mucopolysaccharidosis type III, sanfilippo syndrome". Pediatrician (St. Petersburg). 12 (4): 69–81. doi:10.17816/ped12469-81. ISSN 2587-6252. S2CID 245186102.
- Fedele AO (2015-11-25). "Sanfilippo syndrome: causes, consequences, and treatments". The Application of Clinical Genetics. 8: 269–281. doi:10.2147/TACG.S57672. PMC 4664539. PMID 26648750.
- Jakobkiewicz-Banecka J, Gabig-Ciminska M, Kloska A, Malinowska M, Piotrowska E, Banecka-Majkutewicz Z, et al. (June 2016). "Glycosaminoglycans and mucopolysaccharidosis type III". Frontiers in Bioscience. 21 (7): 1393–1409. doi:10.2741/4463. PMID 27100513.
- Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, et al. (December 2010). "Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations". Annals of Neurology. 68 (6): 876–887. doi:10.1002/ana.22092. PMID 21061399. S2CID 205342620.
- Wagner VF, Northrup H (1993). "Mucopolysaccharidosis Type III". In Adam MP, Everman DB, Mirzaa GM, Pagon RA (eds.). GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 31536183. Retrieved 2023-03-03.
Further reading
- Dietrich CP (January 1969). "Enzymic degradation of heparin. A sulphamidase and a sulphoesterase from Flavobacterium heparinum". The Biochemical Journal. 111 (1): 91–95. doi:10.1042/bj1110091. PMC 1187498. PMID 5775690.
- Mahuran D, Clements P, Hopwood J (June 1983). "A rapid four column purification of 2-deoxy-D-glucoside-2-sulphamate sulphohydrolase from human liver". Biochimica et Biophysica Acta (BBA) - General Subjects. 757 (3): 359–365. doi:10.1016/0304-4165(83)90062-4. PMID 6849981.