Nazarov cyclization reaction
| Nazarov cyclization | |
|---|---|
| Named after | Ivan Nikolaevich Nazarov |
| Reaction type | Ring forming reaction |
| Identifiers | |
| Organic Chemistry Portal | nazarov-cyclization |
| RSC ontology ID | RXNO:0000209 |
The Nazarov cyclization reaction (often referred to as simply the Nazarov cyclization) is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.[1]

As originally described, the Nazarov cyclization involves the activation of a divinyl ketone using a stoichiometric Lewis acid or protic acid promoter. The key step of the reaction mechanism involves a cationic 4π-electrocyclic ring closure which forms the cyclopentenone product (See Mechanism below). As the reaction has been developed, variants involving substrates other than divinyl ketones and promoters other than Lewis acids have been subsumed under the name Nazarov cyclization provided that they follow a similar mechanistic pathway.
The success of the Nazarov cyclization as a tool in organic synthesis stems from the utility and ubiquity of cyclopentenones as both motifs in natural products (including jasmone, the aflatoxins, and a subclass of prostaglandins) and as useful synthetic intermediates for total synthesis. The reaction has been used in several total syntheses and several reviews have been published.[2][3][4][5][6][7]
Mechanism
The mechanism of the classical Nazarov cyclization reaction was first demonstrated experimentally by Charles Shoppee to be an intramolecular electrocyclization and is outlined below. Activation of the ketone by the acid catalyst generates a pentadienyl cation, which undergoes a thermally allowed 4π conrotatory electrocyclization as dictated by the Woodward-Hoffman rules. This generates an oxyallyl cation which undergoes an elimination reaction to lose a β-hydrogen. Subsequent tautomerization of the enolate produces the cyclopentenone product.[8][9]
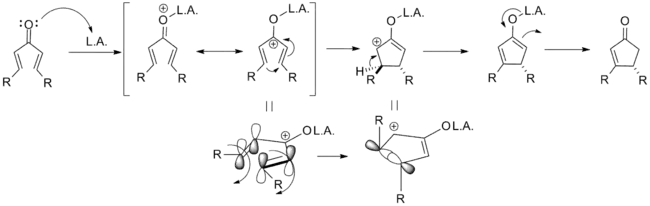
As noted above, variants that deviate from this template are known; what designates a Nazarov cyclization in particular is the generation of the pentadienyl cation followed by electrocyclic ring closure to an oxyallyl cation. In order to achieve this transformation, the molecule must be in the s-trans/s-trans conformation, placing the vinyl groups in an appropriate orientation. The propensity of the system to enter this conformation dramatically influences reaction rate, with α-substituted substrates having an increased population of the requisite conformer due to allylic strain. Coordination of an electron donating α-substituent by the catalyst can likewise increase the reaction rate by enforcing this conformation.[2]
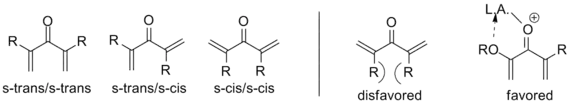
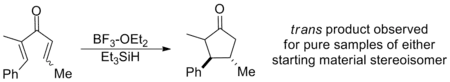
Similarly, β-substitution directed inward restricts the s-trans conformation so severely that E-Z isomerization has been shown to occur in advance of cyclization on a wide range of substrates, yielding the trans cyclopentenone regardless of initial configuration. In this way, the Nazarov cyclization is a rare example of a stereoselective pericyclic reaction, whereas most electrocyclizations are stereospecific. The example below uses triethylsilane to trap the oxyallyl cation so that no elimination occurs.[2] (See Interrupted cyclizations below)
Along this same vein, allenyl vinyl ketones of the type studied extensively by Marcus Tius of the University of Hawaii show dramatic rate acceleration due to the removal of β-hydrogens, obviating a large amount of steric strain in the s-cis conformer.[6]

Classical Nazarov cyclizations
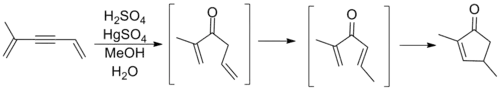
Though cyclizations following the general template above had been observed prior to Nazarov's involvement, it was his study of the rearrangements of allyl vinyl ketones that marked the first major examination of this process. Nazarov correctly reasoned that the allylic olefin isomerized in situ to form a divinyl ketone before ring closure to the cyclopentenone product. The reaction shown below involves an alkyne oxymercuration reaction to generate the requisite ketone.[10]
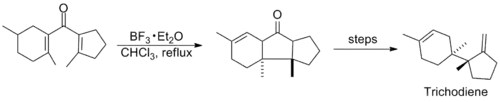
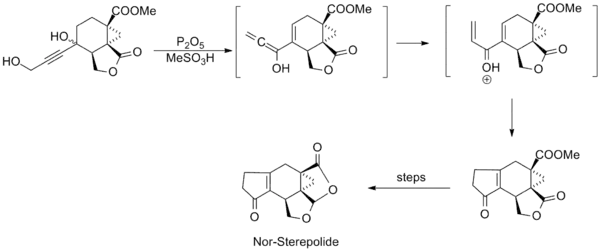
Research involving the reaction was relatively quiet in subsequent years, until in the mid-1980s when several syntheses employing the Nazarov cyclization were published. Shown below are key steps in the syntheses of Trichodiene and Nor-Sterepolide, the latter of which is thought to proceed via an unusual alkyne-allene isomerization that generates the divinyl ketone.[11][12]
Shortcomings
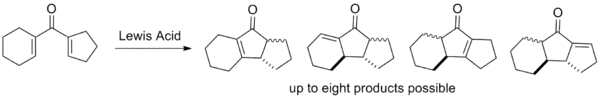
The classical version of the Nazarov cyclization suffers from several drawbacks which modern variants attempt to circumvent. The first two are not evident from the mechanism alone, but are indicative of the barriers to cyclization; the last three stem from selectivity issues relating to elimination and protonation of the intermediate.[2]
- Strong Lewis or protic acids are typically required for the reaction (e.g. TiCl4, BF3, MeSO3H). These promoters are not compatible with sensitive functional groups, limiting the substrate scope.
- Despite the mechanistic possibility for catalysis, multiple equivalents of the promoter are often required in order to effect the reaction. This limits the atom economy of the reaction.
- The elimination step is not regioselective; if multiple β-hydrogens are available for elimination, various products are often observed as mixtures. This is highly undesirable from an efficiency standpoint as arduous separation is typically required.
- Elimination destroys a potential stereocenter, decreasing the potential usefulness of the reaction.
- Protonation of the enolate is sometimes not stereoselective, meaning that products can be formed as mixtures of epimers.
Modern variants
The shortcomings noted above limit the usefulness of the Nazarov cyclization reaction in its canonical form. However, modifications to the reaction focused on remedying its issues continue to be an active area of academic research. In particular, the research has focused on a few key areas: rendering the reaction catalytic in the promoter, effecting the reaction with more mild promoters to improve functional group tolerance, directing the regioselectivity of the elimination step, and improving the overall stereoselectivity. These have been successful to varying degrees.
Additionally, modifications focused on altering the progress of the reaction, either by generating the pentadienyl cation in an unorthodox fashion or by having the oxyallyl cation "intercepted" in various ways. Furthermore, enantioselective variants of various kinds have been developed. The sheer volume of literature on the subject prevents a comprehensive examination of this field; key examples are given below.
Silicon-directed cyclization
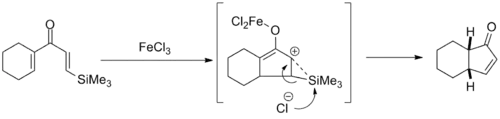
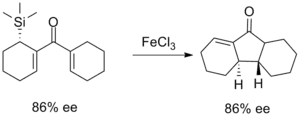
The earliest efforts to improve the selectivity of the Nazarov cyclization took advantage of the β-silicon effect in order to direct the regioselectivity of the elimination step. This chemistry was developed most extensively by Professor Scott Denmark of the University of Illinois, Urbana-Champaign in the mid-1980s and utilizes stoichiometric amounts of iron trichloride to promote the reaction. With bicyclic products, the cis isomer was selected for to varying degrees.[13]
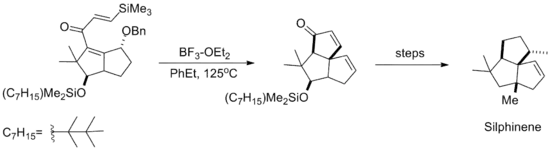
The silicon-directed Nazarov cyclization reaction was subsequently employed in the synthesis of the natural product Silphinene, shown below. The cyclization takes place before elimination of the benzyl alcohol moiety, so that the resulting stereochemistry of the newly formed ring arises from approach of the silyl alkene anti to the ether.[10]
Polarization
Drawing on the substituent effects compiled over various trials of the reaction, Professor Alison Frontier of the University of Rochester developed a paradigm for "polarized" Nazarov cyclizations in which electron donating and electron withdrawing groups are used to improve the overall selectivity of the reaction. Creation of an effective vinyl nucleophile and vinyl electrophile in the substrate allows catalytic activation with copper triflate and regioselective elimination. In addition, the electron withdrawing group increases the acidity of the α-proton, allowing selective formation of the trans-α-epimer via equilibration.[14]
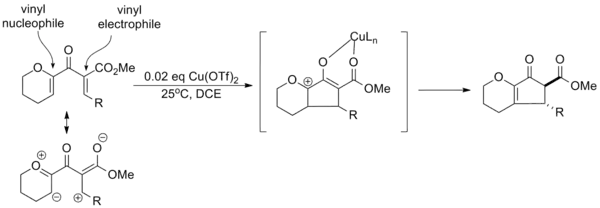
It is often possible to achieve catalytic activation using a donating or withdrawing group alone, although the efficiency of the reaction (yield, reaction time, etc.) is typically lower.
Alternative cation generation
By extension, any pentadienyl cation regardless of its origin is capable of undergoing a Nazarov cyclization. There have been a large number of examples published where the requisite cation is arrived at by a variety of rearrangements.[2] One such example involves the silver catalyzed cationic ring opening of allylic dichloro cylopropanes. The silver salt facilitates loss of chloride via precipitation of insoluble silver chloride.[15]
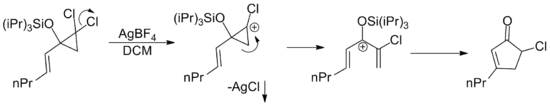
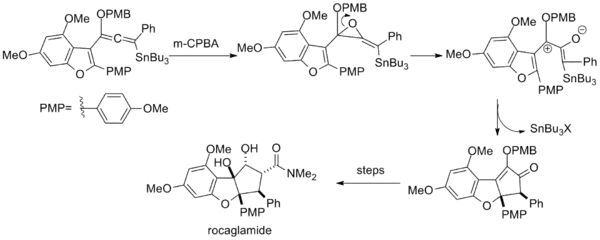
In the total synthesis of rocaglamide, epoxidation of a vinyl alkoxyallenyl stannane likewise generates a pentadienyl cation via ring opening of the resultant epoxide.[16]
Interrupted cyclization
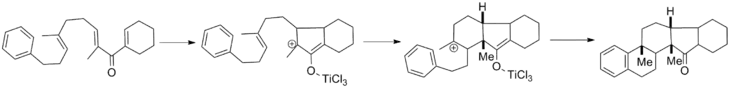
Once the cyclization has occurred, an oxyallyl cation is formed. As discussed extensively above, the typical course for this intermediate is elimination followed by enolate tautomerization. However, these two steps can be interrupted by various nucleophiles and electrophiles, respectively. Oxyallyl cation trapping has been developed extensively by Fredrick G. West of the University of Alberta and his review covers the field.[17] The oxyallyl cation can be trapped with heteroatom and carbon nucleophiles and can also undergo cationic cycloadditions with various tethered partners. Shown below is a cascade reaction in which successive cation trapping generates a pentacyclic core in one step with complete diastereoselectivity.[18]
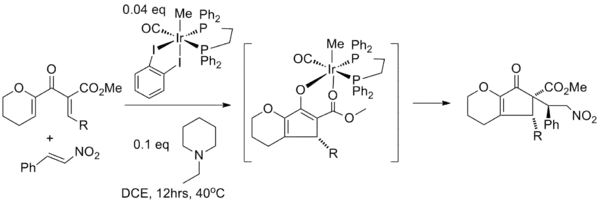
Enolate trapping with various electrophiles is decidedly less common. In one study, the Nazarov cyclization is paired with a Michael reaction using an iridium catalyst to initiate nucleophilic conjugate addition of the enolate to β-nitrostyrene. In this tandem reaction the iridium catalyst is required for both conversions: it acts as the Lewis acid in the Nazarov cyclization and in the next step the nitro group of nitrostyrene first coordinates to iridium in a ligand exchange with the carbonyl ester oxygen atom before the actual Michael addition takes place to the opposite face of the R-group.[19]
Enantioselective variants
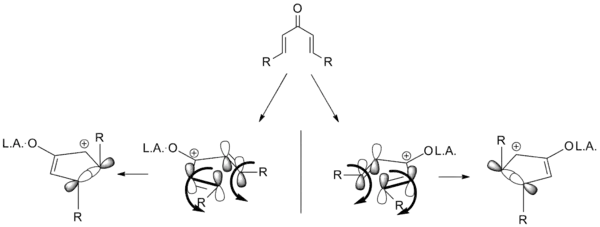
The development of an enantioselective Nazarov cyclization is a desirable addition to the repertoire of Nazarov cyclization reactions. To that end, several variations have been developed utilizing chiral auxiliaries and chiral catalysts. Diastereoselective cyclizations are also known, in which extant stereocenters direct the cyclization. Almost all of the attempts are based on the idea of torquoselectivity; selecting one direction for the vinyl groups to "rotate" in turn sets the stereochemistry as shown below.[2]
Silicon-directed Nazarov cyclizations can exhibit induced diastereoselectivity in this way. In the example below, the silyl-group acts to direct the cyclization by preventing the distant alkene from rotating "towards" it via unfavorable steric interaction. In this way the silicon acts as a traceless auxiliary. (The starting material is not enantiopure but the retention of enantiomeric excess suggests that the auxiliary directs the cyclization.)[2]
Tius's allenyl substrates can exhibit axial to tetrahedral chirality transfer if enantiopure allenes are used. The example below generates a chiral diosphenpol in 64% yield and 95% enantiomeric excess.[2]
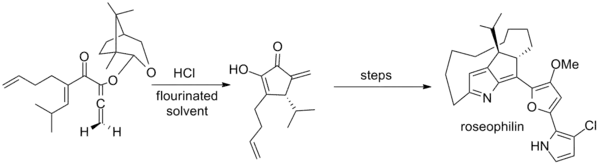
Tius has additionally developed a camphor-based auxiliary for achiral allenes that was employed in the first asymmetric synthesis of roseophilin. The key step employs an unusual mixture of hexafluoro-2-propanol and trifluoroethanol as solvent.[2][20]
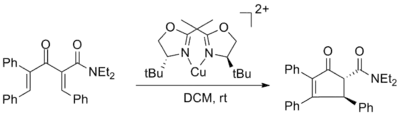
The first chiral Lewis acid promoted asymmetric Nazarov cyclization was reported by Varinder Aggarwal and utilized copper (II) bisoxazoline ligand complexes with up to 98% ee. The enantiomeric excess was unaffected by use of 50 mol% of the copper complex but the yield was significantly decreased.[2]
Related Reactions
Extensions of the Nazarov cyclization are generally also subsumed under the same name. For example, an α-β, γ-δ unsaturated ketone can undergo a similar cationic conrotatory cyclization that is typically referred to as an iso-Nazarov cyclization reaction.[21] Other such extensions have been given similar names, including homo-Nazarov cyclizations and vinylogous Nazarov cyclizations.[22][23]
Retro-Nazarov reaction
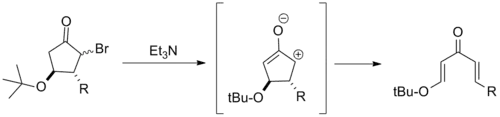
Because they overstabilize the pentadienyl cation, β-electron donating substituents often severely impede Nazarov cyclization. Building from this, several electrocyclic ring openings of β-alkoxy cyclopentanes have been reported. These are typically referred to as retro-Nazarov cyclization reactions.[2]
Imino-Nazarov reaction
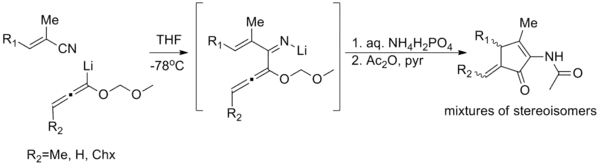
Nitrogen analogues of the Nazarov cyclization reaction (known as imino-Nazarov cyclization reactions) have few instances; there is one example of a generalized imino-Nazarov cyclization reported (shown below),[24] and several iso-imino-Nazarov reactions in the literature.[25][26] Even these tend to suffer from poor stereoselectivity, poor yields, or narrow scope. The difficulty stems from the relative over-stabilization of the pentadienyl cation by electron donation, impeding cyclization.[27]
References
- Nazarov, I.N.; Zaretskaya, I.I. (1941), Izv. Akad. Nauk. SSSR, Ser. Khim: 211–224
{{citation}}: Missing or empty|title=(help) - Frontier, A. J.; Collison, C. (2005), "The Nazarov cyclization in organic synthesis. Recent advances.", Tetrahedron, 61 (32): 7577–7606, doi:10.1016/j.tet.2005.05.019
- Santelli-Rouvier, C.; Santelli, M. (1983), "The Nazarov Cyclization", Synthesis, 1983 (6): 429–442, doi:10.1055/s-1983-30367
- Denmark, S.E.; Habermas, K.L.; Jones, T. K. (1994), "The Nazarov Cyclization", Organic Reactions, 45: 1–158
- Denmark, S.E. (1991), Paquette, L.A. (ed.), "The Nazarov and Related Cationic Cyclizations", Comprehensive Organic Synthesis, Oxford: Pergamon Press, 5: 751–784, doi:10.1016/b978-0-08-052349-1.00138-4, ISBN 9780080523491
- Tius, M. A. (2005), "Some New Nazarov Chemistry", Eur. J. Org. Chem., 2005 (11): 2193–2206, doi:10.1002/ejoc.200500005
- Pellissier, Hélène (2005), "Recent developments in the Nazarov process", Tetrahedron, 61 (27): 6479–6517, doi:10.1016/j.tet.2005.04.014
- Shoppee, C. W.; Lack, R. E. (1969), "Intramolecular electrocyclic reactions. Part I. Structure of 'bromohydroxyphorone': 3-bromo-5-hydroxy-4,4,5,5-tetramethylcyclopent-2-enone", Journal of the Chemical Society C: Organic (10): 1346–1349, doi:10.1039/J39690001346
- Shoppee, C. W.; Cooke, B. J. A. (1972), "Intramolecular electrocyclic reactions. Part II. Reactions of 1,5-di-phenylpenta-1,4-dien-3-one", Journal of the Chemical Society, Perkin Transactions 1: 2271, doi:10.1039/p19720002271
- Kürti, L.; Czakó, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis (1 ed.). Burlington, MA: Elsevier Academic Press. pp. 304–305. ISBN 9780124297852.
- Harding, K. E.; Clement, K. S. (1984), "A Highly Stereoselective, Convergent Synthesis of (±)-Trichodiene", The Journal of Organic Chemistry, 49 (20): 3870–3871, doi:10.1021/jo00194a054
- Arai, Y.; Takeda, K.; Masuda, K.; Koizumi, T. (1985), "Synthesis of(±)-Nor-Sterepolide", Chemistry Letters, 14 (10): 1531–1534, doi:10.1246/cl.1985.1531
- Denmark, S.E.; Jones, T.K. (1982), "Silicon-Directed Nazarov Cyclization", J. Am. Chem. Soc., 104 (9): 2642–2645, doi:10.1021/ja00373a055
- He, W.; Sun, X.; Frontier, A.J. (2003), "Polarizing the Nazarov Cyclization: Efficient Catalysis under Mild Conditions", J. Am. Chem. Soc., 125 (47): 14278–14279, doi:10.1021/ja037910b, PMID 14624567
- Grant, T.N. & West, F.G. (2006), "A New Approach to the Nazarov Reaction via Sequential Electrocyclic Ring Opening and Ring Closure", J. Am. Chem. Soc., 128 (29): 9348–9349, doi:10.1021/ja063421a, PMID 16848467
- Malona, J.A.; Cariou, K.; Frontier, A.J. (2009), "Nazarov Cyclization Initiated by Peracid Oxidation: The Total Synthesis of (±)-Rocaglamide", J. Am. Chem. Soc., 131 (22): 7560–7561, doi:10.1021/ja9029736, PMC 2732401, PMID 19445456
- Grant, T.N.; Rieder, C.J.; West, F.G. (2009), "Interrupting the Nazarov reaction: domino and cascade processes utilizing cyclopentenyl cations", Chem. Commun. (38): 5676–5688, doi:10.1039/B908515G, PMID 19774236
- Bender, J.A.; Arif, A.M.; West, F.G. (1999), "Nazarov-Initiated Diastereoselective Cascade Polycyclization of Aryltrienones", J. Am. Chem. Soc., 121 (32): 7443–7444, doi:10.1021/ja991215f
- Janka, M.; He, W.; Haedicke, I.E.; Fronczek, F.R.; Frontier, A.J.; Eisenberg, R. (2006), "Tandem Nazarov Cyclization-Michael Addition Sequence Catalyzed by an Ir(III) Complex", J. Am. Chem. Soc., 128 (16): 5312–5313, doi:10.1021/ja058772o, PMID 16620081
- Harrington, P.E.; Tius, M.A. (1999), "A Formal Total Synthesis of Roseophilin: Cyclopentannelation Approach to the Macrocyclic Core", Org. Lett., 1 (4): 649–652, doi:10.1021/ol990124k, PMID 10823195
- Jung, M. E.; Yoo, D. (2007), "Unprecedented Rearrangement of a 4-Alkoxy-5-bromoalk-2-en-1-ol to a Cyclopentenone via an Iso-Nazarov Cyclization Process", The Journal of Organic Chemistry, 72 (22): 8565–8568, doi:10.1021/jo071104w, PMID 17910498
- De Simone, F.; Andrès, J.; Torosantucci, R.; Waser, J. r. m. (2009), "Catalytic Formal Homo-Nazarov Cyclization", Organic Letters, 11 (4): 1023–1026, doi:10.1021/ol802970g, PMID 19199774
- Rieder, C. J.; Winberg, K. J.; West, F. G. (2009), "Cyclization of Cross-Conjugated Trienes: The Vinylogous Nazarov Reaction", Journal of the American Chemical Society, 131 (22): 7504–7505, doi:10.1021/ja9023226, PMID 19435345
- Tius, M. A.; Chu, C. C.; Nieves-Colberg, R. (2001), "An imino Nazarov cyclization", Tetrahedron Letters, 42 (13): 2419–2422, doi:10.1016/s0040-4039(01)00201-5
- Kim, S.-H.; Cha, J. K. (2000), "Synthetic Studies Toward caphalotaxine: Functionalization of Tertiary N-Acylhemiaminals by Nazarov Cyclization", Synthesis, 2000 (14): 2113–2116, doi:10.1055/s-2000-8711
- Larini, P.; Guarna, A.; Occhiato, E. G. (2006), "The Lewis Acid-Catalyzed Nazarov Reaction of 2-(N-Methoxycarbonylamino)-1,4-pentadien-3-ones", Org. Lett., 8 (4): 781–784, doi:10.1021/ol053071h, PMID 16468766
- Smith, D. A.; Ulmer, C. W. (1997), "Effects of Substituents in the 3-Position on the [2 + 2] Pentadienyl Cation Electrocyclization", The Journal of Organic Chemistry, 62 (15): 5110–5115, doi:10.1021/jo9703313