Mesoblastic nephroma
Congenital mesoblastic nephroma, while rare, is the most common kidney neoplasm diagnosed in the first three months of life and accounts for 3-5% of all childhood renal neoplasms.[2][3] This neoplasm is generally non-aggressive and amenable to surgical removal. However, a readily identifiable subset of these kidney tumors has a more malignant potential and is capable of causing life-threatening metastases. Congenital mesoblastic nephroma was first named as such in 1967 but was recognized decades before this as fetal renal hamartoma or leiomyomatous renal hamartoma.[4]
| Mesoblastic nephroma | |
|---|---|
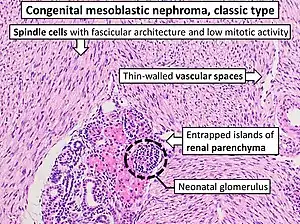 | |
| Congenital mesoblastic nephroma, classic type, with typical features.[1] H&E stain. | |
| Specialty | Oncology, obstetrics and gynaecology, urology |
| Types | Wilms tumor, congenital infantile sarcoma, Rhabdoid tumor, Clear cell sarcoma of the kidney, Infantile myofibromatosis |
| Treatment | surgical removal |
Presentation
Congenital mesoblastic nephroma typically (76% of cases) presents as an abdominal mass which is detected prenatally (16% of cases) by ultrasound or by clinical inspection (84% of cases) either at birth or by 3.8 years of age (median age ~1 month). The neoplasm shows a slight male preference. Concurrent findings include hypertension (19% of cases), polyhydramnios (i.e. excess of amniotic fluid in the amniotic sac) (15%), hematuria (11%), hypercalcemia (4%), and elevated serum levels of the kidney-secreted, hypertension-inducing enzyme, renin (1%). Congenital anomalies have been reported in 11 patients: 6 with genitourinary anomalies, 2 with gastrointestinal anomalies, 1 with hydrocephalus, and 1 with the Beckwith–Wiedemann syndrome. The vast majority of patients present with localized (i.e. non-metastatic) disease.[2][5] Most patients' disease is classified at presentation as stage I or II (i.e. localized), few patients present with stage III (i.e. locally advanced/infiltrating), and virtually no patients present with stage IV (metastases present or V (i.e. tumors in both kidneys) disease (see staging of renal cancer).[2]
Tumor pathology
Congenital Mesoblastic nephroma is a malignant tumorous growth of the kidney's mesenchyme (i.e. connective tissue cells). Histologic examination of these tumors provides critical information on their prognoses. This examination divides congenital mesoblastic nephroma into three types:
- 1) The classic type occurs in ~39% of patients. Its tissues show interlaced spindle-shaped smooth muscle cells evidencing low mitotic activity with no evidence of tumor encapsulation; and infiltration into and entrapment of normal kidney tissue.[2]
- 2) The cellular type occurs in ~42% of patients.[2] Its tissues show densely packed fibrosarcoma-like cells evidencing high rates of mitosis, less infiltration of normal kidney tissue, and multiple areas of hemorrhage and cysts.[2][4]
- 3) The mixed type occurs in ~19% of patients. It shows a mixture of the classic and cellular types in different areas of the neoplasm.[2]
Genetics
A study conducted in 1998 found that congenital mesoblastic nephroma tissues taken from some patients contained an acquired mutation, the ETV6-NTRK3 fusion gene. This gene results from a translocation of genetic material from the ETV6 gene located on the short arm (designated p) of chromosome 12 at position p13.2 (i.e. 12p13.2) to the NTRK3 gene located on the long arm (designated q) of chromosome 15 at position q25.3 (i.e. 15q25.3). This ETV6-NTRK3 gene fusion is notated as t(12;15)(p13;q25) and consists of the 5' end of ETV6 fused to the 3' end of NTRK3.[4] In consequence, the chimeric protein product of this gene lacks ETV6 protein's transcription factor activity while having NTRK3 protein's tyrosine kinase in an unregulated and continuously active form. Either event can drive the malignant growth of cells but in most cases the chimeric protein's tyrosine kinase activity appears responsible for doing so.[6] Based on a limited number of genetic studies (a total of 65 patients), the ETY6-NTRK3 fusion gene appears to occur in most cases of the cellular and some cases of the mixed but no cases of the classical types of congenital mesolastic nephroma.[2][4][7] However, a more recent study of 19 patients detected the fused gene in all 8 cases of cellular, 5 of 6 cases of mixed, and 0 of 5 cases of classic mesoblastic nephroma. This suggests that expression of this fused gene may be more common in cellar and mixed mesoblastic nephroma than previously appreciated.[4]
Trisomy, i.e. pathological presence of an extra chromosome, also occurs in these neoplasms. Trisomy of chromosome 11 (e.g. trisomy 11) appears to be the most commonly found trisomy in this disease, being detected in 7 of 13 genetically studied cases.[2] Individual case reports have also found trisomy 8 (9 cases), 17 (4 cases), 20 (4 cases), 7 (3 cases), 10 (3 cases), 18 (2 cases), 2 (2 cases), and 9 (2 cases) associated with the disease.[2][4] The contribution of these trisomies to the development of mesoblastic nephroma is unclear.
Diagnosis
Diagnosis of mesoblastic nephroma and its particular type (i.e. classic, mixed, or cellular) is made by histological examination of tissues obtained at surgery. Besides its histological appearance, various features of this disease aid in making a differential diagnosis that distinguish it from the following childhood neoplasms:
- Wilms tumor is the most common childhood kidney neoplasm, representing some 85% of cases. Unlike mesoblastic nephroma, <2% of Wilms tumor patients present at under 3 months of age and most present in patients of >3 years of age. Bilateral kidney tumors, concurrent birth defects, and/or metastatic disease at presentation favor a diagnosis of Wilms tumor.[5]
- congenital infantile sarcoma is a rare aggressive sarcoma typically presenting in the lower extremities, head, or neck of infants during their first year of life. The histology, association with the ETV6-NRTK3 fusion gene along with certain chromosome trisomies, and the distribution of markers for cell type (i.e. cyclin D1 and Beta-catenin) within this tumor are the same as those found in cellular mesoblastic nephroma. Mesoblastic nephroma and congenital infantile sarcoma appear to be the same diseases with mesoblastic lymphoma originating in the kidney and congenital infantile sarcoma originating in non-renal tissues.[4][5][8][9]
- Rhabdoid tumor, which accounts for 5-10% of childhood kidney neoplasms, occurs predominantly in children from 1 to 2 years of age. Unlike mesoblastic nephroma, rhabdoid tumors may present with tumors in other tissues including in ~13% of cases, the brain. Rhabdoid tumors have a distinctive histology and abnormalities (i.e. loss of heterozygosity, single nucleotide polymorphism, and deletions) in chromosome 22.[10]
- Clear cell sarcoma of the kidney, which is responsible for 5-10% of childhood pediatric tumors, occurs predominantly in children from 2 to 3 years of age. Unlike mesoblastic nephroma, clear cell sarcoma of the kidney presents with metastasis, particularly to bone, in 5-6% of cases; it histology is diverse and has been mistaken for mesoblastic nephroma. One chromosomal translocations t,(10;17)(q22;p13), has been repeatedly reported to be associated with clear cell sarcoma of the kidney.[11][12]
- Infantile myofibromatosis is a fibrous tumor of infancy and childhood most commonly presenting during the first 2 years of life as a single subcutaneous nodule of the head and neck region or less commonly as multiple lesions of skin, muscle, bone, and in ~33% of these latter cases, visceral organs. All of these lesions have an excellent prognosis and can regress spontaneously except for those in which there is visceral involvement where the prognosis is poor.[13] While infantile myofibromatosis and classic mesoblastic nephroma have been suggested to be the same diseases because of their very similar histology, studies on the distribution of cell-type markers (i.e. cyclin D1 and Beta-catenin) indicate that they have different cellular origins.[4][5]
Treatment
Based on a survey of >800, surgical removal of the entire involved kidney plus the peri-renal fat appeared curative for the majority of all types of mesoblastic nephroma; the patient overall survival rate was 94%. Of the 4% of non-survivors, half were due to surgical or chemotherapeutic treatments. Another 4% of these patients suffered relapses, primarily in the local area of surgery rare cases of relapse due to lung or bone metastasis.. About 60% of these recurrent cases had a complete remission following further treatment. Recurrent disease was treated with a second surgery, radiation, and/or chemotherapy that often vincristine and actinomycin treatment.[2] Removal of the entire afflicted kidney plus the peri-renal fat appears critical to avoiding local recurrences. In general, patients who were older than 3 months of age at diagnosis or had the cellular form of the disease, stage III disease, or involvement of renal lymph nodes had a higher recurrence rate. Among patients with these risk factors, only those with lymph node involvement are recommended for further therapy.[5]
It has been suggested that mesoblastic nephroma patients with lymph node involvement or recurrent disease might benefit by adding the ALK inhibitor, crizotinib, or a tyrosine kinase inhibitor, either larotrectinib or entrectinib, to surgical, radiation, and/or chemotherapy treatment regimens. These drugs inhibit NTRK3's tyrosine kinase activity.[2] Crizotinib has proven useful in treating certain cases of acute lymphoblastic leukemia that are associated with the ETV6-NTRK3 fusion gene while larotrectinib and entrectinib have been useful in treating various cancers (e.g. a metastatic sarcoma, papillary thyroid cancer, non-small-cell lung carcinoma, gastrointestinal stromal tumor, mammary analog secretory carcinoma, and colorectal cancer) that are driven by mutated, overly active tyrosine kinases. Relevant to this issue, a 16-month-old girl with infantile fibrosarcoma harboring the ETV6–NTRK3 fusion gene was successfully treated with larotrectinib.[2][14] The success of these drugs, however, will likely depend on the relative malignancy-promoting roles of ETV6-NTRK3 protein's tyrosine kinase activity, the lose of ETV6-related transcription activity accompanying formation of ETV6-NTRK3 protein, and the various trisomy chromosomes that populate mesoblastic nephroma.
References
- Image by Mikael Häggström, MD. Source for typical features: Ellen D’Hooghe, M.D., Gordan M. Vujanic, M.D., Ph.D. "Congenital mesoblastic nephroma". Pathology Outlines.
{{cite web}}: CS1 maint: multiple names: authors list (link) Last author update: 10 December 2020. Last staff update: 29 November 2021 - Gooskens SL, Houwing ME, Vujanic GM, Dome JS, Diertens T, Coulomb-l'Herminé A, Godzinski J, Pritchard-Jones K, Graf N, van den Heuvel-Eibrink MM (2017). "Congenital mesoblastic nephroma 50 years after its recognition: A narrative review". Pediatric Blood & Cancer. 64 (7): e26437. doi:10.1002/pbc.26437. PMID 28124468. S2CID 22681362.
- Lamb MG, Aldrink JH, O'Brien SH, Yin H, Arnold MA, Ranalli MA (2017). "Renal Tumors in Children Younger Than 12 Months of Age: A 65-Year Single Institution Review". Journal of Pediatric Hematology/Oncology. 39 (2): 103–107. doi:10.1097/MPH.0000000000000698. PMID 27820132. S2CID 40223322.
- El Demellawy D, Cundiff CA, Nasr A, Ozolek JA, Elawabdeh N, Caltharp SA, Masoudian P, Sullivan KJ, de Nanassy J, Shehata BM (2016). "Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement". Pathology. 48 (1): 47–50. doi:10.1016/j.pathol.2015.11.007. PMID 27020209.
- Wang ZP, Li K, Dong KR, Xiao XM, Zheng S (2014). "Congenital mesoblastic nephroma: Clinical analysis of eight cases and a review of the literature". Oncology Letters. 8 (5): 2007–2011. doi:10.3892/ol.2014.2489. PMC 4186628. PMID 25295083.
- Kar A, Gutierrez-Hartmann A (2013). "Molecular mechanisms of ETS transcription factor-mediated tumorigenesis". Critical Reviews in Biochemistry and Molecular Biology. 48 (6): 522–43. doi:10.3109/10409238.2013.838202. PMC 4086824. PMID 24066765.
- Anderson J, Gibson S, Sebire NJ (2006). "Expression of ETV6-NTRK in classical, cellular and mixed subtypes of congenital mesoblastic nephroma". Histopathology. 48 (6): 748–53. doi:10.1111/j.1365-2559.2006.02400.x. PMID 16681692. S2CID 36404121.
- Ud Din N, Minhas K, Shamim MS, Mushtaq N, Fadoo Z (2015). "Congenital (infantile) fibrosarcoma of the scalp: a case series and review of literature". Child's Nervous System. 31 (11): 2145–9. doi:10.1007/s00381-015-2824-1. PMID 26206116. S2CID 25198570.
- Walther C, Nilsson J, von Steyern FV, Wiebe T, Bauer HC, Nord KH, Gisselsson D, Domanski HA, Mandahl N, Mertens F (2013). "Cytogenetic and single nucleotide polymorphism array findings in soft tissue tumors in infants". Cancer Genetics. 206 (7–8): 299–303. doi:10.1016/j.cancergen.2013.06.004. PMID 23938179.
- Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, Perin JC, Xie H, Shaikh TH, Biegel JA (2009). "Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors". Clinical Cancer Research. 15 (6): 1923–30. doi:10.1158/1078-0432.CCR-08-2091. PMC 2668138. PMID 19276269.
- Gooskens SL, Furtwängler R, Vujanic GM, Dome JS, Graf N, van den Heuvel-Eibrink MM (2012). "Clear cell sarcoma of the kidney: a review". European Journal of Cancer. 48 (14): 2219–26. doi:10.1016/j.ejca.2012.04.009. PMID 22579455.
- Alavi S, Khoddami M, Yazdi MK, Dehghanian P, Esteghamati S (2013). "Clear cell sarcoma of the kidney misdiagnosed as mesoblastic nephroma: a case report and review of the literature". ecancermedicalscience. 7: 311. doi:10.3332/ecancer.2013.311. PMC 3634723. PMID 23634181.
- PDQ Pediatric Treatment Editorial Board (2002). "Childhood Soft Tissue Sarcoma Treatment (PDQ®): Health Professional Version". PDQ Cancer Information Summaries. National Cancer Institute (US). PMID 26389361.
- Laetsch TW, Nagasubramanian R, Casanova M (2017). "Targeting NTRK fusions for the treatment of congenital mesoblastic nephroma". Pediatric Blood & Cancer. 65 (1): e26593. doi:10.1002/pbc.26593. PMID 28440051. S2CID 34088251.
External links
- Congenital mesoblastic nephroma entry in the public domain NCI Dictionary of Cancer Terms
![]() This article incorporates public domain material from Dictionary of Cancer Terms. U.S. National Cancer Institute.
This article incorporates public domain material from Dictionary of Cancer Terms. U.S. National Cancer Institute.