Newton-X
Newton-X[1][2] is a general program for molecular dynamics simulations beyond the Born-Oppenheimer approximation. It has been primarily used for simulations of ultrafast processes (femtosecond to picosecond time scale) in photoexcited molecules. It has also been used for simulation of band envelops of absorption and emission spectra.
 | |
 Snapshot of Newton-X main menu. | |
| Developer(s) | M. Barbatti, G. Granucci, M. Ruckenbauer, F. Plasser, R. Crespo-Otero, J. Pittner, M. Persico, H. Lischka |
|---|---|
| Stable release | 2.2
|
| Written in | Perl, Fortran, C |
| Operating system | Linux |
| Website | www |
Newton-X uses the trajectory surface hopping method, a semi-classical approximation in which the nuclei are treated classically by Newtonian dynamics, while the electrons are treated as a quantum subsystem via a local approximation of the Time-dependent Schrödinger Equation. Nonadiabatic effects (the spread of the nuclear wave packet between several states) are recovered by a stochastic algorithm, which allows individual trajectories to change between different potential energy states during the dynamics.
Capabilities
Newton-X is designed as a platform to perform all steps of the nonadiabatic dynamics simulations, from the initial conditions generation, through trajectories computation, to the statistical analysis of the results. It works interfaced to a number of electronic structure programs available for computational chemistry, including Gaussian, Turbomole, Gamess, and Columbus. Its modular development allows to create new interfaces and integrate new methods. Users’ new developments are encouraged and are in due course included into the main branch of the program.
Nonadiabatic couplings, the central quantity in nonadiabatic simulations, can be either provided by a third-party program or computed by Newton-X. When computed by Newton-X, it is done with a numerical approximation based on overlap of electronic wavefunctions obtained in sequential time steps. A local diabatization method is also available to provide couplings in the case of weak nonadiabatic interactions.[3]
Hybrid combination of methods is possible in Newton-X. Forces computed with different methods for different atomic subsets can be linearly combined to generate the final force driving the dynamics. These hybrid forces may, for instance, be combined into the popular electrostatic-embedding quantum-mechanical/molecular-mechanical method (QM/MM). Important options for QM/MM simulations, such as link atoms, boundaries, and thermostats are available as well.
As part of the initial conditions module, Newton-X can simulate absorption, emission, and photoelectron spectra, using the Nuclear Ensemble approach,[4] which provides full spectral widths and absolute intensities.
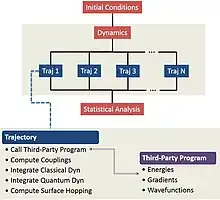
Methods and Interfaces to Third-Party Programs
Newton-X can simulate surface-hopping dynamics with the following programs and quantum-chemical methods:
| Third-Party Program | Methods |
|---|---|
| Columbus | MCSCF, MRCI |
| Turbomole | TDDFT, CC2, ADC(2) |
| Gaussian | MCSCF, TDDFT, TDA, CIS |
| Gamess | MCSCF |
Nonadiabatic couplings
The surface hopping probability depends on the values of the nonadiabatic couplings between electronic states.
Newton-X can either compute nonadiabatic couplings during the dynamics or read them from an interfaced third-party program. The computation of the couplings in Newton-X is done by finite differences, following the Hammes-Schiffer-Tully approach.[5] In this approach, the key quantity for computation of the surface hopping probability, the inner product between the nonadiabatic couplings (τLM) and the nuclear velocities (v) at time t, is given by
,

where the terms are wavefunction overlaps between states L and M in different time steps.

This method can be generally used for any electronic-structure method, provided that a configuration interaction representation of the electronic wavefunction can be worked out. In Newton-X, it is used with a number of quantum-chemical methods, including MCSCF (Multiconfigurational Self-Consistent Field), MRCI (Multi-Reference Configuration Interaction), CC2 (Coupled Cluster to Approximated Second Order), ADC(2) (Algebraic Diagrammatic Construction to Second Order), TDDFT (Time-Dependent Density Functional Theory), and TDA (Tamm-Dankov Approximation). In the case of MCSCF and MRCI, the configuration interaction coefficients are directly used for computation of couplings. For the other methods, the linear-response amplitudes are used as the coefficients of a configuration interaction wavefunction with single excitations.
Spectrum Simulations
Newton-X simulates absorption and emission spectra using the Nuclear Ensemble approach.[4] In this approach, an ensemble of nuclear geometries is built in the initial state and the transition energies and transition moments to the other states are computed for each geometry in the ensemble. A convolution of the results provides spectral widths and absolute intensities.
In the Nuclear Ensemble approach, the photoabsorption cross section for a molecule initially in the ground state and being excited with photoenergy E into Nfs final electronic states is given by
,

where e is the elementary charge, ħ is the reduced Planck constant, m is the electron mass, c is the speed of light, ε0 is the vacuum permittivity, and nr is the refractive index of the medium. The first summation runs over all target states and the second summation runs over all Np points in the nuclear ensemble. Each point in the ensemble has nuclear geometry Rp, transition energy ΔE0,n, and oscillator strength f0,n (for a transition from the ground state into state n). g is a normalized Gaussian function with width δ given by
.

For emission, the differential emission rate is given by
.

In both absorption and emission, the nuclear ensemble can be sampled either from a dynamics simulation or from a Wigner distribution.
Starting from version 2.0, it is possible to use the nuclear ensemble approach to simulate steady and time-resolved photoelectron spectra.
Development and credits
The development of Newton-X started in 2005 at the Institute for the Theoretical Chemistry of the University of Vienna. It was designed by Mario Barbatti in collaboration with Hans Lischka. The original code used and expanded routines written by Giovanni Granucci and Maurizio Persico from the University of Pisa.[2]
A modulus for computation of nonadiabatic couplings based on finite differences of either MCSCF or MRCI wavefunctions was implemented by Jiri Pittner (J. Heyrovsky Institute)[6] and later adapted to work with TDDFT.[7] A modulus for QM/MM dynamics was developed by Matthias Ruckenbauer.[8] Felix Plasser implemented the local diabatization method and dynamics based on CC2 and ADC(2).[3] Rachel Crespo-Otero extended the TDDFT and TDA capabilities.[3] An interface to Gamess was added by Aaron West and Theresa Windus (Iowa State University).[9]
Mario Barbatti coordinates new program developments, their integration into the official version, and the Newton-X distribution.
Distribution and training
Newton-X is distributed free of charges for academic usage and with open source. The original paper[2] describing the program had been cited 190 times by December 22, 2014, according to Google Scholar.
Newton-X counts with a comprehensive documentation and a public discussion forum. A tutorial is also available on line, showing how to use the main features of the program step-by-step. Examples of simulations are shown at a YouTube channel. The program itself is distributed with a collection of input and output files of several worked-out examples.
A number of workshops on nonadiabatic simulations using Newton-X have been organized in Vienna (2008), Rio de Janeiro (2009), Sao Carlos (2011), Chiang Mai (2011, 2015), and Jeddah (2014).[10]
Program philosophy and architecture
A main concept guiding the Newton-X development is that the program should be simple to use, but still providing as many options as possible to customize the jobs. This is achieved by a series of input tools that guide the user through the program options, providing context-dependent variable values always that possible.
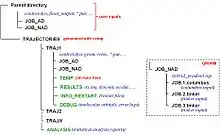
Newton-X is written as a combination of independent programs. The coordinated execution of these programs is done by drivers written in Perl, while the programs dealing with integration of the dynamics and other mathematical aspects are written in Fortran 90 and C. Memory is dynamically allocated and there are no formal limits for most of variables, such as number of atoms or states.
Newton-X works in a three-level parallelization: the first level is a trivial parallelization given by the Independent-Trajectories approach used by the program. Complete sets of input files are redundantly written to allow each trajectory to be executed independently. They can be easily merged for final analysis in a later step. In a second level, Newton-X takes advantage of the parallelization of the third-party programs with which it is interfaced. Thus, a Newton-X simulation using the interface with Gaussian program can be first distributed over a cluster in terms of independent trajectories and each trajectory runs parallelized version of Gaussian. In the third level, the coupling computations in Newton-X are parallelized.
Starting with version (1.3, 2013), Newton-X uses meta-codes to control the dynamics simulation behavior. Based on a series of initial instructions provided by the user, new codes are automatically written and executed on-the-fly. These codes allow, for instance, checking specific conditions to terminate the simulations.
Drawbacks
To keep a modular architecture for easy inclusion of new algorithms, Newton-X is organized as a series of independent programs connected by general program drivers. For this reason, a large amount of input/output is required during the program's execution, reducing its efficiency. When dynamics is based on ab initio methods, this is normally not a problem, as the time bottleneck is in the electronic structure calculation. Low efficiency due to input/output can, however, be relevant with semiempirical methods.
Other problems with the current implementation are the lack of parallelization of the code, especially of the couplings computation, and the restriction of the program to Linux systems.
References
- Barbatti, Mario; Ruckenbauer, Matthias; Plasser, Felix; Pittner, Jiri; Granucci, Giovanni; Persico, Maurizio; Lischka, Hans (January 2014). "Newton-X: a surface-hopping program for nonadiabatic molecular dynamics". Wiley Interdisciplinary Reviews: Computational Molecular Science. 4 (1): 26–33. doi:10.1002/wcms.1158.
- Barbatti, Mario; Granucci, Giovanni; Persico, Maurizio; Ruckenbauer, Matthias; Vazdar, Mario; Eckert-Maksić, Mirjana; Lischka, Hans (August 2007). "The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems". Journal of Photochemistry and Photobiology A: Chemistry. 190 (2–3): 228–240. doi:10.1016/j.jphotochem.2006.12.008.
- Plasser, Felix; Granucci, Giovanni; Pittner, Jiri; Barbatti, Mario; Persico, Maurizio; Lischka, Hans (2012). "Surface hopping dynamics using a locally diabatic formalism: Charge transfer in the ethylene dimer cation and excited state dynamics in the 2-pyridone dimer". The Journal of Chemical Physics. 137 (22): 22A514. Bibcode:2012JChPh.137vA514P. doi:10.1063/1.4738960.
- Crespo-Otero, Rachel; Barbatti, Mario (9 June 2012). "Spectrum simulation and decomposition with nuclear ensemble: formal derivation and application to benzene, furan and 2-phenylfuran". Theoretical Chemistry Accounts. 131 (6). doi:10.1007/s00214-012-1237-4.
- Hammes-Schiffer, Sharon; Tully, John C. (1994). "Proton transfer in solution: Molecular dynamics with quantum transitions". The Journal of Chemical Physics. 101 (6): 4657. Bibcode:1994JChPh.101.4657H. doi:10.1063/1.467455.
- Pittner, Jiri; Lischka, Hans; Barbatti, Mario (February 2009). "Optimization of mixed quantum-classical dynamics: Time-derivative coupling terms and selected couplings". Chemical Physics. 356 (1–3): 147–152. Bibcode:2009CP....356..147P. doi:10.1016/j.chemphys.2008.10.013.
- Barbatti, Mario; Pittner, Jiří; Pederzoli, Marek; Werner, Ute; Mitrić, Roland; Bonačić-Koutecký, Vlasta; Lischka, Hans (September 2010). "Non-adiabatic dynamics of pyrrole: Dependence of deactivation mechanisms on the excitation energy". Chemical Physics. 375 (1): 26–34. Bibcode:2010CP....375...26B. doi:10.1016/j.chemphys.2010.07.014.
- Ruckenbauer, Matthias; Barbatti, Mario; Müller, Thomas; Lischka, Hans (July 2010). "Nonadiabatic Excited-State Dynamics with Hybrid ab Initio Quantum-Mechanical/Molecular-Mechanical Methods: Solvation of the Pentadieniminium Cation in Apolar Media". The Journal of Physical Chemistry A. 114 (25): 6757–6765. Bibcode:2010JPCA..114.6757R. doi:10.1021/jp103101t.
- West, Aaron C.; Barbatti, Mario; Lischka, Hans; Windus, Theresa L. (July 2014). "Nonadiabatic dynamics study of methaniminium with ORMAS: Challenges of incomplete active spaces in dynamics simulations". Computational and Theoretical Chemistry. 1040–1041: 158–166. doi:10.1016/j.comptc.2014.03.015.
- "Newton-X webpage".