Non-canonical base pairing
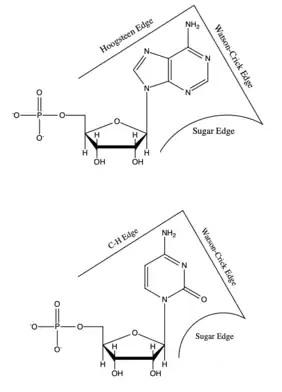
Non-canonical base pairs are planar hydrogen bonded pairs of nucleobases, having hydrogen bonding patterns which differ from the patterns observed in Watson-Crick base pairs, as in the classic double helical DNA. The structures of polynucleotide strands of both DNA and RNA molecules can be understood in terms of sugar-phosphate backbones consisting of phosphodiester-linked D 2’ deoxyribofuranose (D ribofuranose in RNA) sugar moieties, with purine or pyrimidine nucleobases covalently linked to them. Here, the N9 atoms of the purines, guanine and adenine, and the N1 atoms of the pyrimidines, cytosine and thymine (uracil in RNA), respectively, form glycosidic linkages with the C1’ atom of the sugars. These nucleobases can be schematically represented as triangles with one of their vertices linked to the sugar, and the three sides accounting for three edges through which they can form hydrogen bonds with other moieties, including with other nucleobases. The side opposite to the sugar linked vertex is traditionally called the Watson-Crick edge, since they are involved in forming the Watson-Crick base pairs which constitute building blocks of double helical DNA. The two sides adjacent to the sugar-linked vertex are referred to, respectively, as the Sugar and Hoogsteen (C-H for pyrimidines) edges.
Each of the four different nucleobases are characterized by distinct edge-specific distribution patterns of their respective hydrogen bond donor and acceptor atoms, complementarity with which, in turn, define the hydrogen bonding patterns involved in base pairing. The double helical structures of DNA or RNA are generally known to have base pairs between complementary bases, Adenine:Thymine (Adenine:Uracil in RNA) or Guanine:Cytosine. They involve specific hydrogen bonding patterns corresponding to their respective Watson-Crick edges, and are considered as Canonical Base Pairs. At the same time, the helically twisted backbones in the double helical duplex DNA form two grooves, major and minor, through which the hydrogen bond donor and acceptor atoms corresponding respectively to the Hoogsteen and sugar edges are accessible for additional potential molecular recognition events.
Experimental evidences reveal that the nucleotide bases are also capable of forming a wide variety of pairing between bases in various geometries, having hydrogen bonding patterns different from those observed in canonical base pairs. These base pairs, which are generally referred to as Non-Canonical Base Pairs, are held together by multiple hydrogen bonds, and are mostly planar and stable. Most of these play very important roles in shaping the structure and function of different functional RNA molecules. In addition to their occurrences in several double stranded stem regions, most of the loops and bulges that appear in single-stranded RNA secondary structures form recurrent 3D motifs, where non-canonical base pairs play a central role. Non-canonical base pairs also play crucial roles in mediating the tertiary contacts in RNA 3D structures.
History
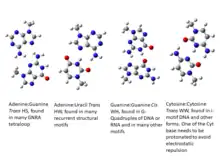
Double helical structures of DNA as well as folded single stranded RNA are now known to be stabilized by Watson-Crick base pairing between the purines, adenine and guanine, with the pyrimidines, thymine (or uracil for RNA) and cytosine. In this scheme, the N1 atoms of the purine residues respectively form hydrogen bond with the N3 atoms of the pyrimidine residues in A:T and G:C complementarity. The second hydrogen bond in A:T base pairs involves the N6 amino group of adenine and the O4 atom of thymine (or uracil in RNA). Similarly, the second hydrogen bond in G:C base pairs involves O6 atom and N4 amino group of guanine and cytosine, respectively. The G:C base pairs also have a third hydrogen bond involving the N2 amino group of guanine and the O2 atom of cytosine. However, even till about twenty years after this scheme was initially proposed by James D. Watson and Francis H.C. Crick,[1] experimental evidences suggesting other forms of base-base interactions continued to draw the attention of researchers investigating the structure of DNA.[2][3] The first high resolution structure of a adenine:thymine base pair, as solved by Karst Hoogsteen by single crystal X-ray crystallography in 1959[4] revealed a structure whose geometry was very different from what was proposed by Watson and Crick. It had two hydrogen bonds involving N7 and N6 atoms of adenine and N3 and O4 (or O2) atoms of thymine. It may be noted that due to use of thymine base with methyl group representing sugar, a symmetry axis appears passing through N1 and C6 atoms and the O2 and O4 atoms appears identical. In order to distinguish this alternate base pairing scheme from the Watson-Crick scheme, base pairs where a hydrogen bond involves the N7 atom of a purine residue have been referred to as Hoogsteen base pair, and later, the purine base edge which includes its N7 atom is referred to as its Hoogsteen edge. The first high resolution structure of guanine:cytosine pair, obtained by W. Guschelbauer also was similar to the Hoogsteen base pair, although this structure required an unusual protonation of N1 imino nitrogen of cytosine, which is possible only at significantly lower pH.[5] Experimental evidences, including low resolution NMR studies[6] as well as high resolution X-ray crystallographic studies,[7] supporting Watson-Crick base pairing were obtained as late as in the early '70s. Almost a decade later, with the advent of efficient DNA synthesis methods, Richard Dickerson[8] followed by several other groups, solved structures of the physiological double helical B-DNA with a complete helical turn, based on the crystals of synthetic DNA oligomers.[9][10][11] The pairing geometries of the A:T (A:U in RNA) and G:C pairs in these structures confirmed the common or canonical form of base pairing as proposed by Watson and Crick, while those with all other geometries, and compositions, are now referred to as non-canonical base pairs.
It was noticed that even in double stranded DNA, where canonical Watson Crick base pairs associate the two complementary anti-parallel strands together, there were occasional occurrences of Hoogsteen and other non-Watson-Crick base pairs.[12][13][14][15][16][17] It was also proposed that within Watson-Crick base pair dominated DNA double helices, Hoogsteen base pair formation could be a transient phenomenon.[17]
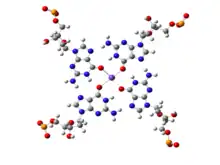
While canonical Watson-Crick base pairs are most prevalent and are commonly observed in a majority of chromosomal DNA and in most functional RNAs, presence of stable non-canonical base pairs is also extremely significant in DNA biology. An example of non-Watson-Crick, or non-canonical, base pairing can be found at the ends of chromosomal DNA. The 3'-ends of chromosomes contain single stranded overhangs with some conserved sequence motifs (such as TTAGGG in most vertebrates). The single stranded region adopts some definite three-dimensional structures, which has been solved by X-ray crystallography as well as by NMR spectroscopy.[18][19][20] The single strands containing the above sequence motifs are found to form interesting four stranded mini-helical structures stabilized by Hoogsteen base pairing between guanine residues. In these structures, four guanine residues form a near planar base quartet, referred to as G-quadruplex, where each guanine participates in base pairing with its neighboring guanine, involving their Watson-Crick and Hoogsteen edges in a cyclic manner. The four central carbonyl groups are often stabilized by potassium ions (K+). From the full genomic sequences of different organisms, it has been observed that telomere like sequences sometimes also interrupt double helical regions near transcription start site of some oncogenes, such as c-myc. It is possible that these sequence stretches form G-quadruplex like structures, which can suppress the expression of the related genes. The complementary cytosine rich sequences, on the other strand, may adopt another similar four stranded structure, the i-motif, stabilized by cytosine:cytosine non-canonical base pairs.
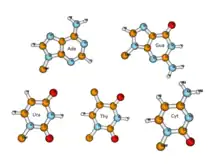
While non-canonical base pairs are still relatively rare in DNA, in RNA molecules, where generally a single polymeric strand folds onto itself to form various secondary and tertiary structures, the occurrence of non-Watson-Crick base pairs turns out to be far more prevalent. As early as in the 1970’s, analysis of the crystal structure of yeast tRNAPhe showed that RNA structures possess significant non-canonical variations in base pairing schemes. Subsequently, the structures of ribozymes, ribosome, riboswitches, etc. have highlighted their abundance, and hence the need for a comprehensive characterization of Non-Canonical Base Pairs. These three-dimensional RNA structures generally possess several secondary structural motifs, such as double helical stems, stems with hairpin loops, symmetric and asymmetric internal loops, kissing loops between two hairpin motifs, pseudoknots, continuous stacks between two segments of helices, multi helix junctions[21][22] etc. along with single stranded regions. These secondary structural motifs, except for the single stranded motifs, are stabilized by hydrogen bonded base pairs and several of these are non-canonical base pairs, including G:U Wobble base pairs.
It is notable in this context, that the Wobble hypothesis of Francis Crick predicted the possibility of G:U base pair, in place of the canonical G:C or A:U base pairs, also mediating the recognition between mRNA codons and tRNA anticodons, during protein synthesis. The G:U wobble base pair is the most numerously observed non-canonical base pair. While, because of its geometric similarity with the canonical base pairs, they frequently occur in the double helical stem regions of RNA structures, the geometric differences continue to draw the attention of nucleic acid researchers, providing new insights related to its structural significance. It may be noted that though the base pairs in the folded RNA structures, give rise to double helical stems, its two cleft regions – the major groove and minor groove, differ in their respective dimensions from those in DNA double helices. Unlike for those in DNA, the sequence discriminating major grooves in RNA double helices are very narrow and deep. On the other hand the minor groove regions, though wide and shallow, do not carry much sequence specific information in terms of the hydrogen bonding donor-acceptor positioning of the corresponding base pair edges.[23] The G:U wobble base pairs, along with the various other non-canonical base pairs, introduce variations in the structures of RNA double helices, thus enhancing the accessibility of the discriminating major groove edges of associated base pairs. This has been seen to be very important for molecular recognition steps during tRNA aminoacylation as well as in ribosome functions.[24]
Considering the immense importance of the non-canonical base pairs in RNA structure, folding and functions, researchers from multiple domains – biology, chemistry, physics, mathematics, computer science, etc., have joined in the effort to understand their structure, dynamics, function and their consequences. The complexities associated with experimental handling of RNA further underline the importance of diverse theoretical inputs towards addressing these issues.
Types
Two bases may approach each other in various ways, eventually leading to specific molecular recognition mediated by, often non-canonical, base pairing interactions, in addition to strong stacking interactions. These are essential for the process of RNA single strands folding into three-dimensional structures. Early studies on such unusual base pairs by Jiri Sponer, Pavel Hobza and their group were somewhat disadvantaged due to the unavailability of suitable unambiguous systematic naming schemes.[25] While some of the observed base pair were assigned names following the Saenger nomenclature scheme.[26] others were arbitrarily assigned names by different researchers. It may be mentioned that some attempts were also made by Michael Levitt and coworkers to classify base-base association in terms of adjacency of bases, through either pairing or stacking interactions.[27] There was clearly a need for a classification scheme for different types of non-canonical base pairs, which could comprehensively and unambiguously handle newer variants coming up due to the rapid increase in the sampling space. Different approaches which have evolved in response to this need are described below.
Based on hydrogen bonding
| Interacting edges | Glycosidic bond orientation | Nomenclature | Local strand direction |
|---|---|---|---|
| Watson-Crick/Watson-Crick | Cis | cWW or cis Watson-Crick/Watson-Crick | Antiparallel |
| Watson-Crick/Watson-Crick | Trans | tWW or trans Watson-Crick/Watson-Crick | Parallel |
| Watson-Crick/Hoogsteen | Cis | cWH or cis Watson-Crick/Hoogsteen | Parallel |
| Watson-Crick/Hoogsteen | Trans | tWH or trans Watson-Crick/Hoogsteen | Antiparallel |
| Watson-Crick/Sugar edge | Cis | cWS or cis Watson-Crick/Sugar edge | Antiparallel |
| Watson-Crick/Sugar edge | Trans | tWS or transWatson-Crick/Sugar edge | Parallel |
| Hoogsteen/Hoogsteen | Cis | cHH or cis Hoogsteen/Hoogsteen | Antiparallel |
| Hoogsteen/Hoogsteen | Trans | tHH or trans Hoogsteen/Hoogsteen | Parallel |
| Hoogsteen/Sugar edge | Cis | cHS or cis Hoogsteen/Sugar edge | Parallel |
| Hoogsteen/Sugar edge | Trans | tHS or trans Hoogsteen/Sugar edge | Antiparallel |
| Sugar edge/Sugar edge | Cis | cSS or cis Sugar edge/Sugar edge | Antiparallel |
| Sugar edge/Sugar edge | Trans | tSS or trans Sugar edge/Sugar edge | parallel |
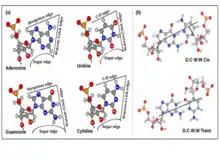
The nucleotide bases are nearly planar heterocyclic moieties, with conjugated pi-electron cloud, and with several hydrogen bonding donors and accepters distributed around the edges, usually designated as W, H or S, based on whether the edges can respectively be involved in forming Watson-Crick base pair, Hoogsteen base pair, or, whether the edge is adjacent to the C2’-OH group of the ribose sugar. Eric Westhof and Neocles Leontis[28] used these edge designations to propose a currently widely accepted nomenclature scheme for base pairs. The hydrogen bonding donor and acceptor atoms could thus be classified in terms of their positioning along their three edges, namely the Watson-Crick or W edge, the Hoogsteen or H edge, and the Sugar or S edge. Since base pairs are mediated through hydrogen bonding interactions based on hydrogen bond donor-acceptor complementarity, this, in turn, provides a convenient bottoms-up approach towards classifying base pair geometries in terms of respective interacting edges of the participating bases. It may be noted that, unlike the Hoogsteen edge of purines, the corresponding edges of the pyrimidine bases do not have any polar hydrogen bond acceptor atom such as N7. However, these bases have C—H groups at their C6 and C5 atoms, which can act as weak hydrogen bond donors, as proposed by Gautam Desiraju.[29] The Hoogsteen edge, hence, is also called Hoogsteen/C-H edge in a unified scheme for designating equivalent positions of purines as well as pyrimidines. Thus, the total number of possible edge combinations involved in base pairing are 6, namely Watson-Crick/Watson-Crick (or W:W), Watson-Crick/Hoogsteen (or W:H), Watson-Crick/Sugar (or W:S), Hoogsteen/Hoogsteen (or H:H), Hoogsteen/Sugar (or H:S) and Sugar/Sugar (or S:S).
In the canonical Watson-Crick base pairs, the glycosidic bonds attaching the N9 (of purine) and N1 (of pyrimidine) of the paired bases with their respective sugar moieties, are on the same side of the mean hydrogen bonding axis, and are hence called Cis Watson-Crick base pairs. However, the relative orientations of the two sugars may also be Trans with respect to the mean hydrogen bonding direction giving rise to a distinct Trans Watson-Crick geometric class, consisting of species which were earlier referred to as reverse Watson-Crick base pairs according to Saenger nomenclature. The possibility of both Cis and Trans glycosidic bond orientation for each of the 6 possible edge combinations, gives rise to 12 geometric families of base pairs (see table).
According to the Leontis-Westhoff scheme, any base pair can be systematically and unambiguously named using the syntax <Base_1: Base_2><Edge_1: Edge_2><Glycosidic Bond Orientation> where Base_1 and Base_2 carry information on respective base identities and their nucleotide number. This nomenclature scheme also allows us to enumerate the total number of distinct possible base pair types. For a given glycosidic bond orientation, say Cis, the four naturally occurring bases each have three possible edges for formation of base pairs giving rise to 12 such possible base pairing edge identities, each of which can in principle form base pairing with any edge of another base, irrespective of complementarity. This gives rise to a 12x12 symmetric matrix displaying 144 pairwise permutations of base pairing edge identities, where, apart from the 12 diagonal entries, others include repeat combinations. Thus, there are 78 (= 12 + 132/2) unique entries corresponding to the cis glycosidic bond orientation. Considering both cis and trans glycosidic bond orientations, the number of base pair types amounts to 156.
Of course, this number 156 is only an indicator. It includes base-edge combinations where base pairs cannot be formed due to absence of hydrogen bond donor acceptor complementarities. For example, potential pairing between two guanine residues utilizing their Watson-Crick edges in cis form (cWW) is not supported by hydrogen bonding donor-acceptor complementarity, and is not observed with consistent hydrogen bonding pattern. This method of enumerating the possible number of distinct base pair types also does not consider possibilities of multimodality or bifurcated base pairs, or even instances of base pairs involving modified bases, protonated bases and water or ion mediation in hydrogen bond formation. Two cytosine bases can form trans Watson-Crick/Watson-Crick (tWW) base pairing with their neutral as well as hemi protonated forms, possibly both, giving rise to the i-motif DNA. However, both C(+):C tWW and C:C tWW, are counted as one type among 156 possible types.
Based on isosteres
Although significant differences are there between structures of non-canonical base pairs belonging to different geometric families, some base pairs within the same geometric family have been found to substitute each other without disrupting the overall structure. These base pairs are called isosteric base pairs. Isosteric base pairs always belong to same geometric families, but all the base pairs in a particular geometric family are not always isosteric. Two base pairs are called isosteric if they meet the following three criteria: (i) The C1′–C1′ distances should be similar; (ii) the paired bases should be related by the similar rotation in 3D space; and (iii) H-bonds formation should occur between equivalent base positions.[30][31] A detailed approach towards quantifying isostericity, in terms of an IsoDiscrepancy Index (IDI), which can facilitate reliable prediction regarding which base pair substitutions can potentially occur in conserved motifs, was formulated by Neocles Leontis, Craig Zirbel and Eric Westhof.[32] Based on IDI values and available base pair structural data, the group maintains a curated online base pair catalogue and an updated set of Isostericity Matrices (IM) corresponding to each of the 12 geometric families. Using this resource, one can comprehensively classify different types of canonical and non-canonical base pairs in terms of their positions in the Isostericity Matrices. This approach, for example, indicates that the four base pair types: A:U cWW, U:A cWW, G:C cWW and C:G cWW are isosteric to each other. Thus, as also confirmed by detailed sequence comparisons, double mutations altering A:U cWW to U:A cWW or even to G:C cWW may not disturb the structure, and, unless stability issues are involved, the function of the related RNA. It was also found that the wobble G:U cWW base pair is not really isosteric to U:G cWW base pair, indicating that such double mutations may significantly affect the functioning of the corresponding RNA. On the other hand, some of the base pairs which are stabilized involving Sugar edge of the bases are mutually isosteric.
Based on local strand direction
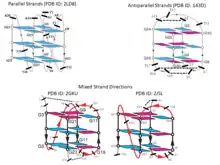
It may be noted here that because of the geometric relationship of the bases with the sugar phosphate backbone, these 12 geometric families of base pairs are associated with two possible local strand orientations, namely parallel and antiparallel. For the 6 families with edge combinations involving Watson-Crick and Sugar edges, W:W, W:S and S:S, cis and trans families are respectively associated with antiparallel and parallel 5' to 3' local strand direction. Introduction of the Hoogsteen edge, as one of the partners in the combination, causes an inversion in the relationship. Thus, for W:H and H:S, cis and trans respectively correspond to parallel and antiparallel local strand orientation. As expected, when both the edges are H, a double inversion is observed, and H:H cis and trans correspond respectively to antiparallel and parallel local strand orientations. The annotation of local strand orientation in terms of parallel and antiparallel directions helps to understand which faces of the individual bases can be seen for a given base pair from the 5’- or the 3’ sides. This annotation also helps in classifying the 12 geometries into two groups of 6 each, where the geometries can potentially interconvert within each group, by in-plane relative rotation of the bases. However, one should note that the above theory is applicable only when the glycosidic torsion angles of both the nucleotide residues are anti. Notably, crystallographic observations[33] and energetic[34] considerations indicate that syn glycosidic torsions are also quite possible. Hence the above classification of parallel or antiparallel nature of strand directions, by itself, does not always provide the complete understanding.
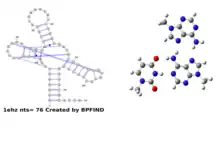
Various functional RNA molecules are stabilized, in their specific folded pattern, by both canonical as well as non-canonical base pairs. Most tRNA molecules, for example, are known to have four short double helical segments, giving rise to a cloverleaf like two-dimensional structure. The three-dimensional structure of tRNA, however, takes an L-shape. This is mediated by several non-canonical base pairs and base triplets. The D-loop and TψC loop are held together by several such base pairs. There is a variety of non-canonical base pair varieties, which can be browsed through different websites such as NDB,[36] RNABPDB,[37] RNABP COGEST,[38] etc., to get a better understanding.
It may be noted that the above scheme is valid for naturally occurring nucleotide bases. However, there are plenty of examples of post-transcriptional chemical modifications of the bases, many of which are seen in tRNAs or ribosomes. It may be important to understand their structural features also.[39][40]
Identification
In case of double helical DNA, identification of base pairs is quite trivial using molecular visualizers such as VMD, RasMol, PyMOL etc. It is, however, not so simple for single stranded folded functional RNA molecules. Several algorithms have been implemented in software tools for the automated detection of base pairs in RNA structures solved by X-ray crystallography, NMR or other methods. Essentially the programs detect hydrogen bonds between two bases, and ensure their (near) planar orientation, before reporting that they constitute a base pair. Since most of the structures of RNA, available in public domain, are solved by X-ray crystallography, the positions of hydrogen atoms are rarely reported. Hence, detection of hydrogen bond becomes a non-trivial job.
The DSSR algorithm[41] by Lu and Wilma K. Olson considers two bases to be paired when they detect one or more hydrogen bond(/s) between the bases, by actually modeling the positions of the hydrogen atoms, and by ensuring the perpendiculars to the two bases being nearly parallel to each other. The positions of the hydrogen atoms can be deduced by converting Internal Coordinates (bond length, bond angle and torsion angle) along with positions of precursor atoms, such as amino group nitrogen atoms and those bonded to the nitrogen or Z-matrix to external Cartesian Coordinates. The base pairs identified by this method are listed in NDB[42] and FR3D[43] databases.
A unique way of identification of base pairs in RNA was incorporated in MC-Annotate[44] by Francois Major. In this algorithm they make use of the positions of the hydrogen atoms as well as lone-pair electrons using suitable molecular mechanics/dynamics force-fields[45] and derive hydrogen bond formation probabilities for them. The final identifications of base pairs are done based on these probabilities and approach of hydrogen atoms to lone-pairs electrons of nitrogen or oxygen. This method also attempted to classify the base pair nomenclature with additional information of each interacting edge, such as Ws indicating the sugar edge corner of the Watson-Crick edge, Wh representing the Hoogsteen edge corner of Watson-Crick edge, Bw indicating bifurcated three-center hydrogen bond involving both the hydrogen atoms of amino groups to form hydrogen bonds with a carbonyl oxygen involving both of its lone-pairs, etc. As claimed by the authors, this nomenclature scheme adds some additional features to the Leontis-Westhof (LW)[28] scheme and may be referred to as the LW+ scheme. A major advantage of this scheme lies in its ability to distinguish between alternative base pairing geometries, where multimodality is observed within an LW family. This method, however, does not consider the possible participation of the 2'-OH group of the ribose sugars in base pair formation.
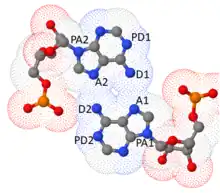
Another algorithm, namely BPFIND by Dhananjay Bhattacharyya and coworkers,[46] demands at least two hydrogen bonds using two distinct sets of donors and acceptors atoms between the bases. This hypothesis driven algorithm considers distances between two pairs of atoms (hydrogen bond donor (D1 and D2) and acceptor (A1 and A2) and four suitably chosen precursor atoms (PD1, PD2, PA1, PA2) corresponding to the D's and A's. Small values of such distances in conjunction with large values of the angles defined by θ1(PD1—D1—A1), θ2(D1—A1—PA1), θ3(PD2—D2—A2), θ4(D2—A2—PA2) (close to 180o or πc) ensures two structural features which characterize well defined base pairs: i) the hydrogen bonds are strong and linear and ii) the two bases are co-planar. Notably, so long as one restricts the search to base pairs which are stabilized by at least two distinct hydrogen bonds, the above algorithms, by and large, yield the same set of base pairs in different RNA structures.
Sometimes in the crystal structures it is observed that two closely spaced bases are oriented in such a way that apart from the regular hydrogen bonds two additional electronegative hydrogen bond acceptor atoms are very close to each other, which may cause electrostatic repulsion. The concept of protonated base pairing, implicating a possible protonation of one of these electronegative, (potentially) hydrogen bond acceptor atoms thus converting it into a hydrogen bond donor, was introduced to explain stability of such geometries.[47][48][46] Some of the NMR derived structures also support the protonation hypothesis, but possibly more rigorous studies using neutron diffraction or other techniques would be able to confirm it. The quality of the crystal structures permitting, some algorithms also attempted to detect water or cation mediated base pair formation.
Stability
The canonical Watson-Crick base pairs, G:C and A:T/U as well as most of the non-canonical ones are stabilized by two or more (e.g. 3 in the case of G:C cWW) hydrogen bonds. Justifiably, a significant amount of research on non-canonical base pairs has been carried out towards bench-marking their strengths (interaction energies) and (geometric) stability against those of the canonical base pairs. It may be noted here that base pair geometries, as observed in the crystal structures, are often influenced by several interactions present in the crystal environment, thus perturbing their intrinsically stable geometries arising out of the hydrogen bonding and related interactions between the two bases. Therefore, in principle, it is possible that the observed geometries in some cases are intrinsically unstable, and that they are stabilized by other interactions provided by the environment. Several groups have attempted to determine the interaction energies in these non-canonical base pairs using different quantum chemistry based approaches, such as Density functional theory (DFT) or MP2 methods.[49][50][51][52][53][54][55][56][57] These methods were applied on suitably truncated, hydrogen-added, and geometry optimized models of the base (or nucleoside) pairs extracted from PDB structures. Depending upon the optimization protocol, typically three types of interaction energies have been reported. In the first method, the base pair model geometries, isolated from their respective environments, are fully optimized without any constraints.[50][52][55][56] thus providing the intrinsic geometries and interaction energies of the isolated models. This procedure, however, sometimes leads to optimized geometries of base pairs involving edges different from initial crystal geometry. Abhijit Mitra and collaborators also used an additional second protocol, where the heavy atom (non-hydrogen) coordinates are retained as in the crystal geometries, optimizing only the positions of the added hydrogen atoms.[51][54][57] In the third protocol, followed mostly by Jiri Sponer and his group,[49] optimization was carried out with constraints on some angles and dihedrals. Given that the models are extracted from their respective crystal structures, and are isolated from their crystal environments, the second and the third protocols provide two different approaches towards approximating the environmental effects, without explicit considerations of any specific environmental interactions. This has further been addressed in some reports by considering specific environmental factors, such as coordination with Magnesium, or even some covalent modifications to the bases.[50]
All the three protocols are useful in their respective contexts. Further, a comparison of the model geometries, obtained by the different protocols, provide an idea regarding both, the stability of the corresponding base pair geometries, as well as regarding the probable extent and nature of environmental influences. It was found that most non-canonical base pairs, having two or more hydrogen bonds, generally maintain the same hydrogen bonding pattern in the crystal and in fully optimized in isolation geometries, respectively, thus indicating their intrinsic geometric stability. Interaction energies calculated from these optimized models also indicated the energetic stability of the corresponding non-canonical base pairs. The previous notion that non-canonical base pairs are weaker than the Watson-Crick base pairs, was found to be incorrect. Interaction energies between the bases of Several base pairs, such as G:G tWW, G:G cWH, A:U cHW, G:A cWW, G:U cWW, etc., are found to be larger than that of canonical A:U cWW base pair.[58]
Of course all non-canonical base pairs are not necessarily very strong or stable in terms of interaction energy. Several base pairs have been detected on the basis of weak hydrogen bonds involving C—H…O/N atoms, where interaction energies are rather small. Further, geometry optimizations of some of the observed base pairs, in particular, but not limited to those involving weak hydrogen bonds, or those stabilized by single hydrogen bonds, were found to adopt alternate geometries,[51][52][57] thus indicating their intrinsic lack of geometric stability. These alteration of hydrogen bonding schemes, giving rise to changes in base pairing family upon free optimization, may have some functional implication in RNA, such as their action as conformational switch. Accordingly, as mentioned above in the Sponer’s protocol, there have been some attempts to restrain the experimentally observed geometry while carrying out geometry optimization[49] for interaction energy calculations. Interestingly, in several cases, interaction energies calculated for these ‘away from intrinsically stable’ geometries also indicate good energetic stability.
Though the energetics and geometric stabilities of different non-canonical base pairs do not show any generalized correlations, analysis of several databases, such as RNABPDB and RNABP COGEST, which catalogue structural and energetic features of some of the observed base pair and their stacks, reveal some interesting general trends.
For example, geometry optimizations of several base pairs involving 2’-OH group of sugar residue resulted in significant alterations from their initial geometry. This is possibly due to flexibility of the sugar puckers and glycosidic torsions. The significantly high interaction energies of protonated base pairs, despite the high energy cost of base protonation, also deserve a special mention in this context. This can mostly be attributed to the additional charge-induced dipole interactions which are associated with protonated base pairs.
Structure
Base pairing
An estimated 60% of bases in structured RNA participate in canonical Watson-Crick base pairs.[59] Base pairing occurs when two bases form hydrogen bonds with each other. These hydrogen bonds can be either polar or non-polar interactions. The polar hydrogen bonds are formed by N-H...O/N and/or O-H...O/N interactions. Non-polar hydrogen bonds are formed between C-H...O/N.[60]
Edge interactions
Each base has three potential edges where it can interact with another base. The Purine bases have 3 edges which are able to hydrogen bond. Those are known as the Watson-Crick edge(WC), the Hoogsteen edge(H), and the Sugar edge(S). Pyrimidine bases also have three hydrogen-bonding edges.[59] Like the purine, there is the Watson-Crick edge(WC) and the Sugar edge(S) but the third edge is referred to as the "C-H" edge(H) on the pyrimidine bases. This C-H edge is sometimes also referred to as the Hoogsteen edge for simplicity. The various edges for the purine and pyrimidine bases are shown in Figure 2.[60]
Besides the three edges of interaction, base pairs can also vary in their cis/trans forms. The cis and trans structures depend on the orientation of the ribose sugar as opposed to the hydrogen bond interaction. These various orientations are shown in Figure 3. Therefore, with the cis/trans forms and the 3 hydrogen bond edges, there are 12 basic types of base pairing geometries which can be found in RNA structures. Those 12 types are WC:WC (cis/trans), WC:HC (cis/trans), WC:S (cis/trans), H:S (cis/trans), H:H (cis/trans), and S:S (cis/trans).
Classification
These 12 types can be further divided into more subgroups which are dependent on the directionality of the glycosidic bonds and steric extensions.[61] With all of the various base pair combinations there are 169 theoretically possible base pair combinations. The actual number of base-pair combinations is lower because some combinations result in non-favorable interactions. This number of possible non-canonical base pairs is still being determined as it depends strongly on base pairing criteria .[62] Understanding base pair configuration is similarly difficult since the pairing is depends on the bases surroundings. These surroundings can consist of adjacent base pairs, adjacent loops, or third interactions (such as a base triple).[63]
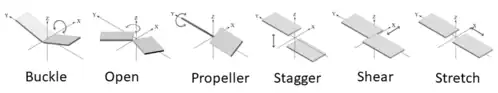
The bonds between various bases are well defined because of their rigid and planar shape. The spatial interactions between the two bases can be classified in 6 rigid-body parameters or intra-base pair parameters (3 translational, 3 rotational) as shown in Figure 4.[64] These parameters describe the base pairs' three dimensional conformation.
The three translational arrangements are known as shear, stretch, and stagger. These three parameters are directly related to the proximity and direction of the hydrogen bonds. The rotational arrangements are buckle, propeller, and opening. Rotational arrangements relate to the non-planar confirmation (as compared to the ideal coplanar geometry).[60] Intra-base pair parameters are used to determine the structure and stabilities of non-canonical base pairs and were originally created for the base pairings in DNA, but were found to also fit the non-canonical base models.[64]
Types
The most common non-canonical base pairs are trans A:G Hoogsteen/sugar edge, A:U Hoogsteen/WC, and G:U Wobble pairs.[65]
Hoogsteen base pairs
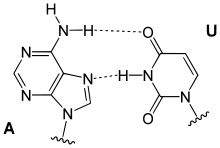
Hoogsteen base pairs occur between adenine (A) and thymine (T); and guanine (G) and cytosine(C); similarly to Watson-Crick base pairs. However, the purine (A and G) takes on an alternative conformation with respect to the pyrimidine. In the A-U Hoogsteen base pair, the adenine is rotated 180° about the glycosidic bond, resulting in an alternative hydrogen bonding scheme which has one hydrogen bond in common with the Watson-Crick base pair (adenine N6 and thymine N4), while the other hydrogen bond, instead of occurring between adenine N1 and thymine N3 as in the Watson-Crick base pair, occurs between adenine N7 and thymine N3.[66] The A-U base pair is shown in Figure 5. In the G-C Watson-Crick base pair, like the A-T Hoogsteen base pair, the purine (guanine) is rotated 180° about the glycosidic bond while the pyrimidine (cytosine) remains in place. One hydrogen bond from the Watson-Crick base pair is maintained (guanine O6 and cytosine N4) and the other occurs between guanine N7 and a protonated cytosine N3 (note that the Hoogsteen G-C base pair has two hydrogen bonds, while the Watson-Crick G-C base pair has three).[66]

Wobble base pairs
Wobble base pairing occur between two nucleotides that are not Watson-Crick base pairs and was proposed by Watson in 1966. The 4 main examples are guanine-uracil (G-U), hypoxanthine-uracil (I-U), hypoxanthine-adenine (I-A), and hypoxanthine-cytosine (I-C). These wobble base pairs are very important in tRNA. Most organisms have less than 45 tRNA molecules even though 61 tRNA molecules would technically be necessary to canonically pair to the codon. Wobble base pairing allows for the 5' anticodon to bond to a non-standard base pair. Examples of wobble base pairs are given in Figure 6.
3-D Structure
The secondary and three-dimensional structures of RNA are formed and stabilized through non-canonical base pairs. Base pairs make up many secondary structural blocks which aid the folding of RNA complexes and three dimensional structures. The overall folded RNA is stabilized by the tertiary and secondary structures canonically base pairing together.[60] Due to the many possible non-canonical base pairs, there are an unlimited amount of structures, which allows for the diverse functions of RNA.[59] The arrangement of the non-canonical bases also allow long-range RNA interactions, recognition of proteins and other molecules, and structural stabilizing elements.[64] Many of the common non-canonical base pairs can be added to a stacked RNA stem without disturbing its helical character.[67]
Secondary
Basic secondary structural elements of RNA include bulges, double helices, hairpin loops, and internal loops. An example of a hairpin loop of RNA is given in Figure 7. As shown in the figure, hairpin loops and internal loops require a sudden change in backbone direction. Non-canonical base pairing allows for the increased flexibility at junctions or turns required in the secondary structure.[60]
Tertiary
Three-dimensional structures are formed through the long-range intra-molecular interactions between the secondary structures. This leads to the formation of pseudoknots, ribose zippers, kissing hairpin loops, or co-axial pseudocontinuous helices.[60] The three-dimensional structures of RNA are primarily determined through molecular simulations or computationally guided measurements.[64] An example of a Pseudoknot is given in Figure 8.
Structural features of a base-pair, formed by two planar rigid units, can be quantified, using six parameters – three translational and three rotational. IUPAC recommended parameters are Propeller, Buckle, Open Angle, Stagger, Shear and Stretch (Figure 8).[68] There are several publicly available software, such as Curves[69] by Richard Lavery, 3DNA[70] by Olson, NUPARM[71][72] by Manju Bansal, etc., which may be used to calculate these parameters. While the first two calculate the parameters of canonical and non-canonical base-pairs relative to the standard canonical Watson-Crick base pairs geometry, the NUPARM algorithm calculates in absolute terms using base pairing edge specific axis system. Hence, for most non-canonical base-pairs, which involve non-Watson-Crick edges, some of the parameters (Open, Shear and Stretch) calculated by Curves or 3DNA are usually large even in their respective intrinsically most stable geometries. On the other hand, the values provided by NUPARM indicate the quality of hydrogen bonding and planarity of the two bases in a more realistic fashion. Thus, the NUPARM Stretch values, indicating separation of the two bases of a base pair, and which depend on optimal hydrogen bonding distances, are always around 3Ǻ. Some other general trends observed in the values of the above parameters may be of interest to note. Most of the cis base pairs are seen to have Propeller values around -10o and small values of Buckle and Stagger. The Open and Shear values often depend on positions of the hydrogen bonding atoms. As for example, GU cWW wobble base pairs have Shear value around -2.2Ǻ while GC or AU cWW base pairs have Shear values around zero. The Open values for most base pairs are close to zero but the values are often rather large for those involving 2’-OH group of sugar in the NUPARM derived parameter set. The trans base pairs, however, do not show any systematic trend in their Propeller values.
Roles
In RNA
The structural hierarchy in RNA is usually described in terms of a stem-loop 2D secondary structure, which further folds to form its 3D tertiary structure, stabilized by what are referred to as long range tertiary contacts. Most often the non-canonical base pairs are involved in those tertiary contacts or extra-stem base pairs. For example, some of the non-canonical base pairs in tRNA appear between the D-stem and TψC loops (Figure 5), which are close in the three-dimensional structure. Such base pairing interactions give stability to the L-shaped structure of tRNA. In this region, some base pairs are found to be additionally hydrogen bonded to a third base. Thus, the 23rd residue is simultaneously paired to 9th and 12th residues, together forming a base triple, the smallest member of the class of higher order multiplets.
Multiplets
One base, in addition to forming proper planar base pairing with a second base, can often participate in base pair formation with a third base forming a base triple. One such classic example is in formation of DNA triple helix, where two bases of two antiparallel strands form consecutive Watson-Crick base pairs in a double helix and a base of a third strand form Hoogsteen base pairing with the purine bases of the Watson-Crick base pairs. Many different types of base triples have been reported in the available RNA structures and have been elegantly classified in the literature.[73] Multiplets are however not limited to triplet formation. Four bases giving rise to a base quartet is now well documented in the structure of the G-quadruplex characteristically found in the telomere. Here four Guanine residues pair up within themselves in a cyclic form involving Watson-Crick/Hoogsteen cis (cWH) base pairing scheme and each of the Guanine bases are found to be respectively interact with two other guanine bases. Three to four such base G-quadruplexes stack on top of the other to form a four stranded DNA structure. In addition to such a cyclic topology, several other topologies of base:base pairings are possible for higher order multiplets such as quartets, pentets etc.[74]
Double helical regions
Non-canonical base pairs quite frequently appear within double helical regions of RNA. The G:U cWW non-canonical base pairs are seen very frequently within double helical regions as this base pair is nearly isosteric to the other canonical ones.[75] Due to complication of strand direction, as elaborated in the Classification section (Table 1), not all types of non-canonical base pairs can be accommodated within double helical regions with anti glycosidic torsion angles. However, many non-canonical base pairs, e.g. A:G tHS (trans Hoogsteen/Sugar edge) or A:U tHW (trans Hoogsteen/Watson-Crick), A:G cWW, etc., are often seen within double helical regions giving rise to symmetric internal loop like motifs. Attempts have been made to classify all such situations where two base pairs (canonical or non-canonical) stack in anti-parallel sense possibly giving rise to double helical regions in RNA structures. These base pairs are quite stable, and they are able to maintain the helical property quite well. The backbone torsion angles around these residues are also generally within reasonable limits: C3'-endo sugar pucker with anti glycosidic torsion, α/γ torsion angles around -60o/60o, β/ε torsion angles around 180o.
Recurrent structural motifs
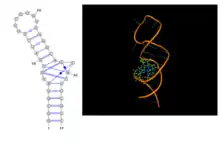
Non-canonical base pairs often appear in different structural motifs, including pseudoknots, with their special hydrogen bonding features. Structural features of these recurrent motifs have been archived in searchable databases, such as, FR3D[76] and RNA FRABASE.[77] Also, several of these motifs can be identified in a given query PDB file by the NASSAM[78] web-server. They are most frequently detected at the termini of double helical segment acting as capping residues, often preceding hairpin loops. The most frequently found non-canonical base pair, namely G:A tSH, is an integral part of GNRA tetraloops, where N can be any nucleotide residue and R is a purine residue. This motif shows some amount of flexibility and alterations of structural features depending on whether the Guanine and Adenine are paired or not. Several other types of tetraloops motifs, such as UNCG, YNMG, GNAC, CUYG, (where Y stands for pyrimidine and M is either Adenine or Cytosine) etc., have been found in available RNA structures. However, these do not generally show involvement of non-canonical base pairing. In addition to these common hairpin motifs, where the loop residues largely remain unpaired, there are also a few motifs where the loop residues make extensive interactions between themselves or with other residues external to the loop. A common example is the C-loop motif,[79][80] where the bulging loop residues make non-canonical base pairing with the bases of double helical regions forming non-canonical base pairing (Figure 9). The extra base pairs in these cases give rise to additional stabilization to the composite double helix containing motif. Non-canonical base pairs are also involved in receptor-loop interaction, such as in T-loop motif.[79] Another interesting example of the involvement of non-canonical base pairs in recurrent contexts was detected as the GAAA receptor motif, which consists of A:A cHS base pair followed by U:A tWH base pair stacked on both sides by G:C cWW base pairs. Here we have successive non-canonical base pairs within an antiparallel RNA double helical domain. Similarly there is an A:A cSH base pair involving two consecutive residues in this motif. Such pairing between consecutive residues, which is also termed as a dinucleotide platform motif, is quite commonly observed. They appear in many RNA structures and the pairing can also be between other bases. Such dinucleotide platform was reported in A:A, A:G, A:U, G:A, G:U base pairs belonging to the cSH class and also in A:A cHH base pairs. These motifs can alter the strand direction within a double helix by formation of kinks. Such dinucleotide platform along with triplet formation is also an integral component of the Sarcin-ricin motif.[81]
Modeling
Prediction of biomolecular structure from sequence alone is a long term goal of scientists working in the fields of bioinformatics, computational chemistry, statistical physics as well as in computer science. Prediction of protein structures from amino acid sequence by methods like homology modeling, comparative modeling, threading, etc were largely successful due to availability of about 1200 unique protein folds. Inspired by the protein experience, there are now several approaches towards predicting RNA structures, albeit with varying degrees of success.
It can be seen that most of the approaches are essentially limited to the prediction of RNA 2D stem-loop structure, also referred to as RNA secondary structure. For example, minimum computed free energy prediction of double helical regions of RNA sequences from the energy of base pairing and stacking interactions, essentially computationally derived from experimental thermodynamic data, was initially introduced by Ruth Nussinov and later by Michael Zuker. This, in turn, has inspired several related modified algorithms, including data on neighboring group interactions etc.[75] Most of these approaches, however, mainly consider data on canonical base pairing, with only a few which also consider thermodynamic data on Hoogsteen base pairs. Thus, in addition to the computational costs and complications associated with the identification of pseudoknots, all these methods also suffer from the drawback associated with the paucity of experimental data on non-canonical base pairs.
However, there are also several approaches which attempt at predicting the tertiary 3D structure corresponding to given predicted 2D structures. There are also a few involving 3D fragment based modeling,[82] which are getting further facilitated with the increasing availability of motif wise curated RNA 3D structure data.[75] It is also encouraging to note that there are now some software and servers, such as MC-Fold,[83] RNAPDBee,[84] RNAWolfe,[85] etc. available for exploring non-canonical base pairing in RNA 3D structures. Some of these methods depend on structural database of RNA, such as FRABASE,[77] to obtain 3D coordinates of motifs containing non-canonical base pairs and stitch the information with 3D structure of double helices containing canonical base pairs.
It may be relevant in this context, to mention about the approach towards 3D model building of double helical regions with both canonical and non-canonical base pairs used in 3DNA by Olson or in RNAHelix[86] by Bhattacharyya and Bansal. These software suites use base pair parameters to generate 3D coordinates of individual dinucleotide steps, which can be extended to model double helices of arbitrary lengths with canonical or non-canonical base pairs.
The above mentioned methods attempt to model a single structure (2D or 3D) of a given RNA sequence. However, growing evidences indicate that a given RNA sequence can adopt ensemble of structures and possibly interconvert between them.[87] This ensembles obviously adopt different base pairing patterns between different sets of residues.[88] Thus, there are enough pointers to suggest that the focus on modeling single structures appears to have been a bottleneck for accurate modeling of RNA structure.
The theoretical prediction of RNA 2D structure and consequently 3D structure can also be confirmed by different chemical probing methods. One of the latest such tools is SHAPE (Selective 2′-hydroxyl acylation analyzed by primer extension), and SHAPE-Directed RNA Secondary Structure Prediction[89] appears to be most promising. Coupled with mutational profiling, ensembles of RNA structures, which often include non-canonical base pairing, can be experimentally studied using the SHAPE-MaP approach.[90] One of the ways ahead today appears to be an integration of Zuker’s minimum free energy approach with experimentally derived SHAPE data, including simulated SHAPE data as outlined in Montaseri et al. (2016)[91] and Spasic et al (2017).[92]
See also
References
![]() This article was adapted from the following source under a CC BY 4.0 license (2023) (reviewer reports):
Dhananjay Bhattacharyya; Abhijit Mitra (8 April 2023). "Non-canonical base pairing" (PDF). WikiJournal of Science. 6 (1): 2. doi:10.15347/WJS/2023.002. ISSN 2470-6345. Wikidata Q39049436.
This article was adapted from the following source under a CC BY 4.0 license (2023) (reviewer reports):
Dhananjay Bhattacharyya; Abhijit Mitra (8 April 2023). "Non-canonical base pairing" (PDF). WikiJournal of Science. 6 (1): 2. doi:10.15347/WJS/2023.002. ISSN 2470-6345. Wikidata Q39049436.
- Watson JD, Crick FH (April 1953). "Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid". Nature. 171 (4356): 737–738. Bibcode:1953Natur.171..737W. doi:10.1038/171737a0. PMID 13054692. S2CID 4253007.
- Nikolova EN, Zhou H, Gottardo FL, Alvey HS, Kimsey IJ, Al-Hashimi HM (December 2013). "A historical account of Hoogsteen base-pairs in duplex DNA". Biopolymers. 99 (12): 955–968. doi:10.1002/bip.22334. PMC 3844552. PMID 23818176.
- Westhof E, Fritsch V (March 2000). "RNA folding: beyond Watson-Crick pairs". Structure. 8 (3): R55–R65. doi:10.1016/s0969-2126(00)00112-x. PMID 10745012.
- Hoogsteen K (1959-10-10). "The structure of crystals containing a hydrogen-bonded complex of 1-methylthymine and 9-methyladenine". Acta Crystallographica. 12 (10): 822–823. doi:10.1107/s0365110x59002389. ISSN 0365-110X.
- Courtois Y, Fromageot P, Guschlbauer W (December 1968). "Protonated polynucleotide structures. 3. An optical rotatory dispersion study of the protonation of DNA". European Journal of Biochemistry. 6 (4): 493–501. doi:10.1111/j.1432-1033.1968.tb00472.x. PMID 5701966.
- Patel DJ, Tonelli AE (October 1974). "Assignment of the proton Nmr chemical shifts of the T-N3H and G-N1H proton resonances in isolated AT and GC Watson-Crick base pairs in double-stranded deoxy oligonucleotides in aqueous solution". Biopolymers. 13 (10): 1943–1964. doi:10.1002/bip.1974.360131003. PMID 4433696. S2CID 39908141.
- Seeman NC, Rosenberg JM, Suddath FL, Kim JJ, Rich A (June 1976). "RNA double-helical fragments at atomic resolution. I. The crystal and molecular structure of sodium adenylyl-3',5'-uridine hexahydrate". Journal of Molecular Biology. 104 (1): 109–144. doi:10.1016/0022-2836(76)90005-x. PMID 957429.
- Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE (1981-05-21). "Structure of a B-DNA dodecamer: Conformation and dynamics". Proceedings of the National Academy of Sciences of the United States of America. 78 (4): 2179–2183. Bibcode:1981PNAS...78.2179D. doi:10.1073/pnas.78.4.2179. PMC 319307. PMID 6941276.
- Wang A, Fujii S, Van Boom J, Van Der Marel G, Van Boeckel S, Rich A (1993). "Molecular Structure of R(GCG)D(TATACGC): A DNA-RNA Hybrid Helix Joined to Double Helical DNA". doi:10.2210/pdb1d96/pdb.
{{cite journal}}: Cite journal requires|journal=(help) - Heinemann U, Alings C (November 1989). "Crystallographic study of one turn of G/C-rich B-DNA". Journal of Molecular Biology. 210 (2): 369–381. doi:10.1016/0022-2836(89)90337-9. PMID 2600970.
- Dock-Bregeon AC, Chevrier B, Podjarny A, Johnson J, de Bear JS, Gough GR, et al. (October 1989). "Crystallographic structure of an RNA helix: [U(UA)6A]2". Journal of Molecular Biology. 209 (3): 459–474. doi:10.1016/0022-2836(89)90010-7. PMID 2479753.
- Patikoglou GA, Kim JL, Sun L, Yang SH, Kodadek T, Burley SK (December 1999). "TATA element recognition by the TATA box-binding protein has been conserved throughout evolution". Genes & Development. 13 (24): 3217–3230. doi:10.1101/gad.13.24.3217. PMC 317201. PMID 10617571.
- Aishima J, Gitti R, Noah J, Gan H, Schlick T, Wolberger C (2002-12-11). "MATalpha2 Homeodomain Bound to DNA". Worldwide Protein Data Bank. doi:10.2210/pdb1k61/pdb. Retrieved 2019-12-17.
- Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK (July 2004). "Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing". Nature. 430 (6997): 377–380. Bibcode:2004Natur.430..377N. doi:10.1038/nature02692. PMID 15254543. S2CID 4379848.
- Kitayner M, Rozenberg H, Rohs R, Suad O, Rabinovich D, Honig B, Shakked Z (April 2010). "Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs". Nature Structural & Molecular Biology. 17 (4): 423–429. doi:10.1038/nsmb.1800. PMC 3280089. PMID 20364130.
- Ethayathulla A, Tse P, Nguyen S, Viadiu H (2012). "Structure of p73 DNA binding domain tetramer modulates p73 transactivation". Proceedings of the National Academy of Sciences of the United States of America. 109 (16): 6066–6071. doi:10.2210/pdb3vd2/pdb. PMC 3341074. PMID 22474346.
- Xu Y, McSally J, Andricioaei I, Al-Hashimi HM (April 2018). "Modulation of Hoogsteen dynamics on DNA recognition". Nature Communications. 9 (1): 1473. Bibcode:2018NatCo...9.1473X. doi:10.1038/s41467-018-03516-1. PMC 5902632. PMID 29662229.
- Parkinson GN, Lee MP, Neidle S (June 2002). "Crystal structure of parallel quadruplexes from human telomeric DNA". Nature. 417 (6891): 876–880. Bibcode:2002Natur.417..876P. doi:10.1038/nature755. PMID 12050675. S2CID 4422211.
- Luu KN, Phan AT, Kuryavyi V, Lacroix L, Patel DJ (August 2006). "Structure of the human telomere in K+ solution: an intramolecular (3 + 1) G-quadruplex scaffold". Journal of the American Chemical Society. 128 (30): 9963–9970. doi:10.1021/ja062791w. PMC 4692383. PMID 16866556.
- Phan AT, Kuryavyi V, Luu KN, Patel DJ (2007-09-25). "Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution". Nucleic Acids Research. 35 (19): 6517–6525. doi:10.1093/nar/gkm706. PMC 2095816. PMID 17895279.
- Hendrix DK, Brenner SE, Holbrook SR (August 2005). "RNA structural motifs: building blocks of a modular biomolecule". Quarterly Reviews of Biophysics. 38 (3): 221–243. doi:10.1017/s0033583506004215. PMID 16817983. S2CID 2632921.
- Laing C, Jung S, Iqbal A, Schlick T (October 2009). "Tertiary motifs revealed in analyses of higher-order RNA junctions". Journal of Molecular Biology. 393 (1): 67–82. doi:10.1016/j.jmb.2009.07.089. PMC 3174529. PMID 19660472.
- Halder S, Bhattacharyya D (November 2013). "RNA structure and dynamics: a base pairing perspective". Progress in Biophysics and Molecular Biology. 113 (2): 264–283. doi:10.1016/j.pbiomolbio.2013.07.003. PMID 23891726.
- Ananth P, Goldsmith G, Yathindra N (August 2013). "An innate twist between Crick's wobble and Watson-Crick base pairs". RNA. 19 (8): 1038–1053. doi:10.1261/rna.036905.112. PMC 3708525. PMID 23861536.
- Šponer J, Leszczynski J, Hobza P (January 1996). "Structures and Energies of Hydrogen-Bonded DNA Base Pairs. A Nonempirical Study with Inclusion of Electron Correlation". The Journal of Physical Chemistry. 100 (5): 1965–1974. doi:10.1021/jp952760f. ISSN 0022-3654.
- Saenger W (1984). Principles of Nucleic Acid Structure. Springer Advanced Texts in Chemistry. New York, NY: Springer New York. pp. 1–8. doi:10.1007/978-1-4612-5190-3. ISBN 978-0-387-90761-1.
- Sykes MT, Levitt M (August 2005). "Describing RNA structure by libraries of clustered nucleotide doublets". Journal of Molecular Biology. 351 (1): 26–38. doi:10.1016/j.jmb.2005.06.024. PMC 2746451. PMID 15993894.
- Leontis NB, Westhof E (April 2001). "Geometric nomenclature and classification of RNA base pairs". RNA. 7 (4): 499–512. doi:10.1017/S1355838201002515. PMC 1370104. PMID 11345429.
- Stombaugh J, Zirbel CL, Westhof E, Leontis NB (April 2009). "Frequency and isostericity of RNA base pairs". Nucleic Acids Research. 37 (7): 2294–2312. doi:10.1093/nar/gkp011. PMC 2673412. PMID 19240142.
- Leontis NB, Stombaugh J, Westhof E (August 2002). "The non-Watson-Crick base pairs and their associated isostericity matrices". Nucleic Acids Research. 30 (16): 3497–3531. doi:10.1093/nar/gkf481. PMC 134247. PMID 12177293.
- Nasalean L, Stombaugh J, Zirbel CL, Leontis NB (2009). "RNA 3D Structural Motifs: Definition, Identification, Annotation, and Database Searching". In Walter NG, Woodson SA, Batey RT (eds.). Non-Protein Coding RNAs. Springer Series in Biophysics. Vol. 13. Berlin, Heidelberg: Springer Berlin Heidelberg. pp. 1–26. doi:10.1007/978-3-540-70840-7_1. ISBN 978-3-540-70833-9.
- Stombaugh J, Zirbel CL, Westhof E, Leontis NB (April 2009). "Frequency and isostericity of RNA base pairs". Nucleic Acids Research. 37 (7): 2294–2312. doi:10.1093/nar/gkp011. PMC 2673412. PMID 19240142.
- Sokoloski JE, Godfrey SA, Dombrowski SE, Bevilacqua PC (October 2011). "Prevalence of syn nucleobases in the active sites of functional RNAs". RNA. 17 (10): 1775–1787. doi:10.1261/rna.2759911. PMC 3185911. PMID 21873463.
- Reichert J, Sühnel J (January 2002). "The IMB Jena Image Library of Biological Macromolecules: 2002 update". Nucleic Acids Research. 30 (1): 253–254. doi:10.1093/nar/30.1.253. PMC 99077. PMID 11752308.
- Darty K, Denise A, Ponty Y (August 2009). "VARNA: Interactive drawing and editing of the RNA secondary structure". Bioinformatics. 25 (15): 1974–1975. doi:10.1093/bioinformatics/btp250. PMC 2712331. PMID 19398448.
- "RNA Basepair Catalog". ndbserver.rutgers.edu. Retrieved 2019-12-17.
- "RNA Base Pair Database(RNABPDB)". hdrnas.saha.ac.in. Retrieved 2019-12-17.
- Bhattacharya S, Mittal S, Panigrahi S, Sharma P, Preethi SP, Paul R, et al. (January 2015). "RNABP COGEST: a resource for investigating functional RNAs". Database. 2015. doi:10.1093/database/bav011. PMC 4360618. PMID 25776022.
- Chawla M, Oliva R, Bujnicki JM, Cavallo L (August 2015). "An atlas of RNA base pairs involving modified nucleobases with optimal geometries and accurate energies". Nucleic Acids Research. 43 (14): 6714–6729. doi:10.1093/nar/gkv606. PMC 4538814. PMID 26117545.
- Seelam PP, Sharma P, Mitra A (June 2017). "Structural landscape of base pairs containing post-transcriptional modifications in RNA". RNA. 23 (6): 847–859. doi:10.1261/rna.060749.117. PMC 5435857. PMID 28341704.
- Lu XJ, Olson WK (September 2003). "3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures". Nucleic Acids Research. 31 (17): 5108–5121. doi:10.1093/nar/gkg680. PMC 212791. PMID 12930962.
- Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, et al. (September 1992). "The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids". Biophysical Journal. 63 (3): 751–759. Bibcode:1992BpJ....63..751B. doi:10.1016/s0006-3495(92)81649-1. PMC 1262208. PMID 1384741.
- Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB (January 2008). "FR3D: finding local and composite recurrent structural motifs in RNA 3D structures". Journal of Mathematical Biology. 56 (1–2): 215–252. doi:10.1007/s00285-007-0110-x. PMC 2837920. PMID 17694311.
- Lemieux S, Major F (October 2002). "RNA canonical and non-canonical base pairing types: a recognition method and complete repertoire". Nucleic Acids Research. 30 (19): 4250–4263. doi:10.1093/nar/gkf540. PMC 140540. PMID 12364604.
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, et al. (May 1995). "A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules". Journal of the American Chemical Society. 117 (19): 5179–5197. doi:10.1021/ja00124a002. ISSN 0002-7863.
- Das J, Mukherjee S, Mitra A, Bhattacharyya D (October 2006). "Non-canonical base pairs and higher order structures in nucleic acids: crystal structure database analysis". Journal of Biomolecular Structure & Dynamics. 24 (2): 149–161. doi:10.1080/07391102.2006.10507108. PMID 16928138. S2CID 21066195.
- Chawla M, Sharma P, Halder S, Bhattacharyya D, Mitra A (February 2011). "Protonation of base pairs in RNA: context analysis and quantum chemical investigations of their geometries and stabilities". The Journal of Physical Chemistry B. 115 (6): 1469–1484. doi:10.1021/jp106848h. PMID 21254753.
- Kelly RE, Lee YJ, Kantorovich LN (June 2005). "Homopairing possibilities of the DNA base adenine". The Journal of Physical Chemistry B. 109 (24): 11933–11939. doi:10.1021/jp050962y. PMID 16852470.
- Sponer JE, Leszczynski J, Sychrovský V, Sponer J (October 2005). "Sugar edge/sugar edge base pairs in RNA: stabilities and structures from quantum chemical calculations". The Journal of Physical Chemistry B. 109 (39): 18680–18689. doi:10.1021/jp053379q. PMID 16853403.
- Oliva R, Cavallo L, Tramontano A (2006-02-06). "Accurate energies of hydrogen bonded nucleic acid base pairs and triplets in tRNA tertiary interactions". Nucleic Acids Research. 34 (3): 865–879. doi:10.1093/nar/gkj491. PMC 1361619. PMID 16461956.
- Bhattacharyya D, Koripella SC, Mitra A, Rajendran VB, Sinha B (August 2007). "Theoretical analysis of noncanonical base pairing interactions in RNA molecules". Journal of Biosciences. 32 (5): 809–825. doi:10.1007/s12038-007-0082-4. PMID 17914224. S2CID 10937466.
- Roy A, Panigrahi S, Bhattacharyya M, Bhattacharyya D (March 2008). "Structure, stability, and dynamics of canonical and noncanonical base pairs: quantum chemical studies". The Journal of Physical Chemistry B. 112 (12): 3786–3796. doi:10.1021/jp076921e. PMID 18318519.
- Sharma P, Mitra A, Sharma S, Singh H, Bhattacharyya D (June 2008). "Quantum chemical studies of structures and binding in noncanonical RNA base pairs: the trans Watson-Crick:Watson-Crick family". Journal of Biomolecular Structure & Dynamics. 25 (6): 709–732. doi:10.1080/07391102.2008.10507216. PMID 18399704. S2CID 13471262.
- Sharma P, Sponer JE, Sponer J, Sharma S, Bhattacharyya D, Mitra A (March 2010). "On the role of the cis Hoogsteen:sugar-edge family of base pairs in platforms and triplets-quantum chemical insights into RNA structural biology". The Journal of Physical Chemistry B. 114 (9): 3307–3320. doi:10.1021/jp910226e. PMID 20163171.
- Brovarets' OO, Yurenko YP, Hovorun DM (2013-06-03). "Intermolecular CH···O/N H-bonds in the biologically important pairs of natural nucleobases: a thorough quantum-chemical study". Journal of Biomolecular Structure & Dynamics. 32 (6): 993–1022. doi:10.1080/07391102.2013.799439. PMID 23730732. S2CID 205574305.
- Marino T (June 2014). "DFT investigation of the mismatched base pairs (T-Hg-T)3, (U-Hg-U)3, d(T-Hg-T)2, and d(U-Hg-U)2". Journal of Molecular Modeling. 20 (6): 2303. doi:10.1007/s00894-014-2303-8. PMID 24878806. S2CID 1986713.
- Mládek A, Sharma P, Mitra A, Bhattacharyya D, Sponer J, Sponer JE (February 2009). "Trans Hoogsteen/sugar edge base pairing in RNA. Structures, energies, and stabilities from quantum chemical calculations". The Journal of Physical Chemistry B. 113 (6): 1743–1755. doi:10.1021/jp808357m. PMID 19152254.
- Mukherjee D, Maiti S, Gouda PK, Sharma R, Roy P, Bhattacharyya D (September 2022). "RNABPDB: Molecular Modeling of RNA Structure-From Base Pair Analysis in Crystals to Structure Prediction". Interdisciplinary Sciences, Computational Life Sciences. 14 (3): 759–774. doi:10.1007/s12539-022-00528-w. PMID 35705797. S2CID 249709744.
- Leontis NB, Westhof E (April 2001). "Geometric nomenclature and classification of RNA base pairs". RNA. 7 (4): 499–512. doi:10.1017/S1355838201002515. PMC 1370104. PMID 11345429.
- Halder S, Bhattacharyya D (November 2013). "RNA structure and dynamics: a base pairing perspective". Progress in Biophysics and Molecular Biology. 113 (2): 264–83. doi:10.1016/j.pbiomolbio.2013.07.003. PMID 23891726.
- Sponer JE, Leszczynski J, Sychrovský V, Sponer J (October 2005). "Sugar edge/sugar edge base pairs in RNA: stabilities and structures from quantum chemical calculations". The Journal of Physical Chemistry B. 109 (39): 18680–9. doi:10.1021/jp053379q. PMID 16853403.
- Sharma P, Sponer JE, Sponer J, Sharma S, Bhattacharyya D, Mitra A (March 2010). "On the role of the cis Hoogsteen:sugar-edge family of base pairs in platforms and triplets-quantum chemical insights into RNA structural biology". The Journal of Physical Chemistry B. 114 (9): 3307–20. doi:10.1021/jp910226e. PMID 20163171.
- Heus HA, Hilbers CW (October 2003). "Structures of non-canonical tandem base pairs in RNA helices: review". Nucleosides, Nucleotides & Nucleic Acids. 22 (5–8): 559–71. doi:10.1081/NCN-120021955. PMID 14565230. S2CID 23265089.
- Olson WK, Li S, Kaukonen T, Colasanti AV, Xin Y, Lu XJ (May 2019). "Effects of Noncanonical Base Pairing on RNA Folding: Structural Context and Spatial Arrangements of G·A Pairs". Biochemistry. 58 (20): 2474–2487. doi:10.1021/acs.biochem.9b00122. PMC 6729125. PMID 31008589.
- Roy A, Panigrahi S, Bhattacharyya M, Bhattacharyya D (March 2008). "Structure, stability, and dynamics of canonical and noncanonical base pairs: quantum chemical studies". The Journal of Physical Chemistry B. 112 (12): 3786–96. doi:10.1021/jp076921e. PMID 18318519.
- Nikolova EN, Zhou H, Gottardo FL, Alvey HS, Kimsey IJ, Al-Hashimi HM (December 2013). "A historical account of Hoogsteen base-pairs in duplex DNA". Biopolymers. 99 (12): 955–68. doi:10.1002/bip.22334. PMC 3844552. PMID 23818176.
- Hermann T, Westhof E (December 1999). "Non-Watson-Crick base pairs in RNA-protein recognition" (PDF). Chemistry & Biology. 6 (12): R335-43. doi:10.1016/s1074-5521(00)80003-4. PMID 10631510.
- Dickerson RE (March 1989). "Definitions and nomenclature of nucleic acid structure components". Nucleic Acids Research. 17 (5): 1797–1803. doi:10.1093/nar/17.5.1797. PMC 317523. PMID 2928107.
- Blanchet C, Pasi M, Zakrzewska K, Lavery R (July 2011). "CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures". Nucleic Acids Research. 39 (Web Server issue): W68–W73. doi:10.1093/nar/gkr316. PMC 3125750. PMID 21558323.
- Lu XJ, Olson WK (July 2008). "3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures". Nature Protocols. 3 (7): 1213–1227. doi:10.1038/nprot.2008.104. PMC 3065354. PMID 18600227.
- Bansal M, Bhattacharyya D, Ravi B (June 1995). "NUPARM and NUCGEN: software for analysis and generation of sequence dependent nucleic acid structures". Computer Applications in the Biosciences. 11 (3): 281–287. doi:10.1093/bioinformatics/11.3.281. PMID 7583696.
- Mukherjee S, Bansal M, Bhattacharyya D (2006-11-24). "Conformational specificity of non-canonical base pairs and higher order structures in nucleic acids: crystal structure database analysis". Journal of Computer-Aided Molecular Design. 20 (10–11): 629–645. Bibcode:2006JCAMD..20..629M. doi:10.1007/s10822-006-9083-x. PMID 17124630. S2CID 9275640.
- Abu Almakarem AS, Petrov AI, Stombaugh J, Zirbel CL, Leontis NB (February 2012). "Comprehensive survey and geometric classification of base triples in RNA structures". Nucleic Acids Research. 40 (4): 1407–1423. doi:10.1093/nar/gkr810. PMC 3287178. PMID 22053086.
- Bhattacharya S, Jhunjhunwala A, Halder A, Bhattacharyya D, Mitra A (May 2019). "Going beyond base-pairs: topology-based characterization of base-multiplets in RNA". RNA. 25 (5): 573–589. doi:10.1261/rna.068551.118. PMC 6467009. PMID 30792229.
- Tabei Y, Tsuda K, Kin T, Asai K (July 2006). "SCARNA: fast and accurate structural alignment of RNA sequences by matching fixed-length stem fragments". Bioinformatics. 22 (14): 1723–1729. doi:10.1093/bioinformatics/btl177. PMID 16690634.
- Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB (January 2008). "FR3D: finding local and composite recurrent structural motifs in RNA 3D structures". Journal of Mathematical Biology. 56 (1–2): 215–252. doi:10.1007/s00285-007-0110-x. PMC 2837920. PMID 17694311.
- Popenda M, Szachniuk M, Blazewicz M, Wasik S, Burke EK, Blazewicz J, Adamiak RW (May 2010). "RNA FRABASE 2.0: an advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures". BMC Bioinformatics. 11 (1): 231. doi:10.1186/1471-2105-11-231. PMC 2873543. PMID 20459631.
- Hamdani HY, Appasamy SD, Willett P, Artymiuk PJ, Firdaus-Raih M (July 2012). "NASSAM: a server to search for and annotate tertiary interactions and motifs in three-dimensional structures of complex RNA molecules". Nucleic Acids Research. 40 (Web Server issue): W35–W41. doi:10.1093/nar/gks513. PMC 3394293. PMID 22661578.
- Petrov AI, Zirbel CL, Leontis NB (October 2013). "Automated classification of RNA 3D motifs and the RNA 3D Motif Atlas". RNA. 19 (10): 1327–1340. doi:10.1261/rna.039438.113. PMC 3854523. PMID 23970545.
- "RNA 3D Motif Atlas". rna.bgsu.edu. Retrieved 2019-12-17.
- Szewczak AA, Moore PB (March 1995). "The sarcin/ricin loop, a modular RNA". Journal of Molecular Biology. 247 (1): 81–98. doi:10.1006/jmbi.1994.0124. PMID 7897662.
- Baulin E, Yacovlev V, Khachko D, Spirin S, Roytberg M (2016). "URS DataBase: universe of RNA structures and their motifs". Database. 2016: baw085. doi:10.1093/database/baw085. PMC 4885603. PMID 27242032.
- Parisien M, Major F (March 2008). "The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data". Nature. 452 (7183): 51–55. Bibcode:2008Natur.452...51P. doi:10.1038/nature06684. PMID 18322526. S2CID 4415777.
- Zok T, Antczak M, Zurkowski M, Popenda M, Blazewicz J, Adamiak RW, Szachniuk M (July 2018). "RNApdbee 2.0: multifunctional tool for RNA structure annotation". Nucleic Acids Research. 46 (W1): W30–W35. doi:10.1093/nar/gky314. PMC 6031003. PMID 29718468.
- Rybarczyk A, Szostak N, Antczak M, Zok T, Popenda M, Adamiak R, et al. (September 2015). "New in silico approach to assessing RNA secondary structures with non-canonical base pairs". BMC Bioinformatics. 16 (1): 276. doi:10.1186/s12859-015-0718-6. PMC 4557229. PMID 26329823.
- Bhattacharyya D, Halder S, Basu S, Mukherjee D, Kumar P, Bansal M (February 2017). "RNAHelix: computational modeling of nucleic acid structures with Watson-Crick and non-canonical base pairs". Journal of Computer-Aided Molecular Design. 31 (2): 219–235. Bibcode:2017JCAMD..31..219B. doi:10.1007/s10822-016-0007-0. PMID 28102461. S2CID 356097.
- Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL (February 2009). "A stress-responsive RNA switch regulates VEGFA expression". Nature. 457 (7231): 915–919. Bibcode:2009Natur.457..915R. doi:10.1038/nature07598. PMC 2858559. PMID 19098893.
- Cruz JA, Westhof E (February 2009). "The dynamic landscapes of RNA architecture". Cell. 136 (4): 604–609. doi:10.1016/j.cell.2009.02.003. PMID 19239882. S2CID 10174208.
- Low JT, Weeks KM (October 2010). "SHAPE-directed RNA secondary structure prediction". Methods. 52 (2): 150–158. doi:10.1016/j.ymeth.2010.06.007. PMC 2941709. PMID 20554050.
- Siegfried NA, Busan S, Rice GM, Nelson JA, Weeks KM (September 2014). "RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP)". Nature Methods. 11 (9): 959–965. doi:10.1038/nmeth.3029. PMC 4259394. PMID 25028896.
- Montaseri S, Ganjtabesh M, Zare-Mirakabad F (2016-11-28). "Evolutionary Algorithm for RNA Secondary Structure Prediction Based on Simulated SHAPE Data". PLOS ONE. 11 (11): e0166965. Bibcode:2016PLoSO..1166965M. doi:10.1371/journal.pone.0166965. PMC 5125645. PMID 27893832.
- Spasic A, Assmann SM, Bevilacqua PC, Mathews DH (January 2018). "Modeling RNA secondary structure folding ensembles using SHAPE mapping data". Nucleic Acids Research. 46 (1): 314–323. doi:10.1093/nar/gkx1057. PMC 5758915. PMID 29177466.