Noonan syndrome
Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations.[1] Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw.[1] Heart problems may include pulmonary valve stenosis.[1] The breast bone may either protrude or be sunken, while the spine may be abnormally curved.[1] Intelligence is often normal.[1] Complications of NS can include leukemia.[1]
| Noonan syndrome | |
|---|---|
| Other names | Male Turner syndrome, Noonan–Ehmke syndrome, Turner-like syndrome, Ullrich–Noonan syndrome[1] |
 | |
| A 12-year-old girl with Noonan syndrome, displaying typical webbed neck and double structural curve with rib deformity. | |
| Specialty | Medical genetics, pediatrics |
| Symptoms | Mildly unusual facial features, short height, congenital heart disease, bleeding problems, skeletal malformations[1] |
| Complications | Leukemia[1] |
| Usual onset | Present at birth[2] |
| Types | Type 1 to 6[3] |
| Causes | Genetic mutation (autosomal dominant)[1] |
| Diagnostic method | Suspected based on symptoms, confirmed with genetic testing[4][2] |
| Differential diagnosis | Cardiofaciocutaneous syndrome, Turner syndrome, Costello syndrome, neurofibromatosis type 1[2][3] |
| Treatment | Based on the symptoms[3] |
| Medication | Growth hormone[3] |
| Prognosis | Depends on the severity of heart problems[3] |
| Frequency | 1 in 1000 (1 in 2,000 severe disease)[4] |
| Named after | Jacqueline Noonan |
A number of genetic mutations can result in Noonan syndrome.[1] The condition may be inherited as an autosomal dominant condition or occur as a new mutation.[3][1] Noonan syndrome is a type of RASopathy, the underlying mechanism for which involves attenuation of the RAS/MAPK cell signaling pathway.[1] The diagnosis may be suspected based on symptoms, medical imaging, and blood tests.[2][4] Confirmation may be achieved with genetic testing.[2]
No cure for NS is known.[5] Treatment is based on the symptoms and underlying problems, and extra support in school may be required.[3] Growth hormone therapy during childhood can increase an affected person's final height.[3] Long-term outcomes typically depend on the severity of heart problems.[3]
An estimated 1 in 1000 people are mildly affected by NS, while about 1 in 2,000 have a more severe form of the condition.[4] Males appear to be affected more often than females.[2] The condition was named after American pediatric cardiologist Jacqueline Noonan, who described her first case in 1963.[2]
Signs and symptoms


The most common signs leading to the diagnosis of Noonan syndrome are unique facial characteristics and musculoskeletal features. The facial characteristics are most prominent in infancy, becoming less apparent with age in many people with Noonan syndrome.[7]
Head
Some of the characteristic features of Noonan syndrome include a large head with excess skin on the back of the neck, low hairline at the nape of the neck, high hairline at the front of the head, triangular face shape, broad forehead, and a short, webbed neck.
In the eyes, hypertelorism (widely set eyes) is a defining characteristic, present in 95% of people with Noonan syndrome. This may be accompanied by epicanthal folds (extra fold of skin at the inner corner of the eye), ptosis (drooping of the eyelids), proptosis (bulging eyes), strabismus (inward or outward turning of the eyes), nystagmus (jerking movement of the eyes) and refractive visual errors.
The nose may be small, wide, and upturned.
The development of the ears and auditory system may be affected in people with Noonan's syndrome. This can result in low-set ears (in over 90%), backward-rotated ears (over 90%), thick helix (outer rim) of ear (over 90%), incomplete folding of ears, chronic otitis media (ear infections), and hearing loss.
Development of the mouth may also be affected in Noonan syndrome. This can result in deeply grooved philtrum (top lip line) (over 90%), micrognathia (undersized lower jaw), high arched palate, articulation difficulties (teeth don't line up) which can lead to dental problems. Similar to the muscular manifestations above, in the mouth, poor tongue control may be observed.
Skin
Skin signs and symptoms in Noonan syndrome include lymphedema (lymph swelling of the extremities), keloid formation, excessive scar formation, hyperkeratosis (overdevelopment of outer skin layer), pigmented nevi (darkly pigmented skin spots), and connective tissue disease.
Musculoskeletal
Abnormalities in the limbs and extremities may occur in Noonan syndrome. This may manifest as bluntly ended fingers, extra padding on fingers and toes, edema of the back of hands and tops of feet, and cubitus valgus (wide carrying angle of the elbows).
For short stature, growth hormone is sometimes combined with IGF-1 (or as an alternative, IGF-1 as a stand-alone) can be used to achieve an increased height/final height quicker. The final adult height of individuals with Noonan syndrome is about 161–167 cm in males and 150–155 cm in females, which approaches the lower limit of normal.[8]
Spinal abnormalities may be present up to 30% of the time and this may require surgery to correct in over 60% of these cases. Other musculoskeletal manifestations in Noonan syndrome are associated with undifferentiated connective-tissue disorders which can be associated with joint contractures (tightness) or joint hypermobility (looseness). Additional factors may present in the form of winging of the scapula, scoliosis, breast bone prominence (pectus carinatum), breast bone depression (pectus excavatum). Muscle abnormalities may present as hypotonia (low muscle tone), which may lead to lordosis (increased hollow in the back) due to poor abdominal muscle tone.
Heart
Noonan syndrome is the second most common syndromic cause of congenital heart disease. This includes pulmonary valvular stenosis (50–60%), atrial septal defects (10–25%), ventricular septal defects (5–20%) and hypertrophic cardiomyopathy (12–35%).
Lungs
Restrictive lung function has been reported in some people.
Gastrointestinal
A number of diverse gastrointestinal (GI) symptoms have been associated with Noonan syndrome. These include swallowing difficulties, low gut motility, gastroparesis (delayed gastric emptying), intestinal malrotation, and frequent or forceful vomiting. These digestive issues may lead to decreased appetite, failure to thrive from infancy to puberty (75%), and occasionally the need for a feeding tube.
Genitourinary system
In some males with Noonan syndrome, testicles do not descend (cryptorchidism).
Circulation
Lymphatic anomalies including Posterior cervical hygroma (webbed neck) and Lymphedema may present in people with Noonan syndrome.
A number of bleeding disorders have been associated with Noonan syndrome, these include platelet dysfunction, Blood clotting disorders, partial deficiency of factor VIII:C, partial deficiency of factor XI:C, partial deficiency of factor XII:C, and an imbalance of plasminogen activator inhibitor type-1 (PAI-1) and tissue plasminogen activator (t-PA) activity. It has been associated with Von Willebrand disease, Amegakaryocytic thrombocytopenia (low platelet count), prolonged activated partial thromboplastin time, combined coagulation defects. When present, these Noonan-syndrome accompanying disorders can be associated with a predisposition to bruise easily, or hemorrhage.
Neurological
Occasionally, Chiari malformation (type 1), may occur, which can lead to hydrocephalus. Seizures have also been reported.
Causes
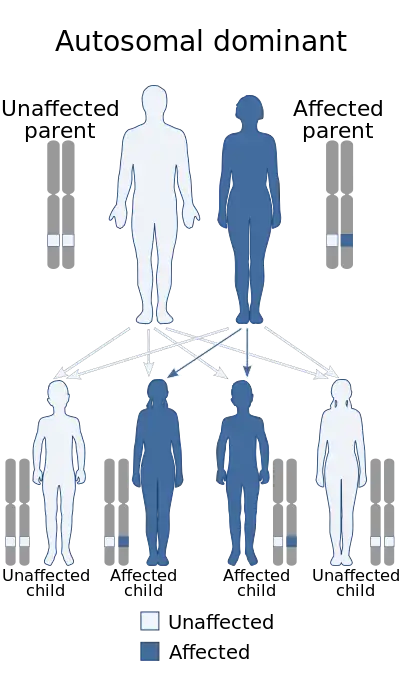
Recurrence in siblings and apparent transmission from parent to child has long suggested a genetic defect with autosomal dominant inheritance and variable expression. Mutations in the Ras/mitogen activated protein kinase signaling pathways are known to be responsible for about 70% of NS cases.[9]
Persons with NS have up to a 50% chance of transmitting it to their offspring. The fact that an affected parent is not always identified for children with NS suggests several possibilities:
- Manifestations could be so subtle as to go unrecognized (variable expressivity)
- NS is heterogeneous, comprising more than one similar condition of differing causes, and some of these may not be inherited.
- A high proportion of cases may represent new, sporadic mutations.
| Type | Online Mendelian Inheritance in Man database | Gene | Year found | Locus | % of cases | Description | Refs. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | The PTPN11 gene encodes the protein tyrosine phosphatase SHP-2. This protein is a component of several intracellular signal transduction pathways involved in embryonic development that modulate cell division, differentiation, and migration, including one mediated by the epidermal growth factor receptor, which is important in the formation of the semilunar heart valves. Duplication of the chromosome region containing PTPN11 can also result in NS. |
[10] [11] |
| NS2 | 605275 | Unknown; autosomal recessive | [12] | ||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | [13] | |
| NS4 | 610733 | SOS1 | 2006 | 2p22 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development.[14] | [15] |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3–17% | [16] |
Heterozygous mutations in NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, and CBL have also been associated with a smaller percentage of NS and related phenotypes.[17]
A condition known as "neurofibromatosis–Noonan syndrome" is associated with neurofibromin.[18]
Diagnosis
NS can be confirmed genetically by the presence of any of the known mutations listed above. However, despite identification of 14 causative genes, the absence of a known mutation will not exclude the diagnosis, as more, as-yet-undiscovered genes can cause NS. Thus, the diagnosis of NS is still based on clinical features. In other words, it is made when a physician feels that a person has enough of the features to warrant the label. The principal values of making a genetic diagnosis are that it guides additional medical and developmental evaluations, it excludes other possible explanations for the features, and it allows more accurate recurrence risk estimates. With more genotype-phenotype correlation studies being performed, a positive genetic diagnosis will help the clinician to be aware of possible anomalies specific to that certain gene mutation. For example, an increase in hypertrophic cardiomyopathy is seen in people with a mutation of KRAS and an increased risk of juvenile myelomonocytic leukemia exists for a mutation of PTPN11. In the future, studies may lead to a targeted management of NS symptoms that depends on what genetic mutation a person has.
Before birth
Prenatal features that might lead physicians to consider a diagnosis of Noonan syndrome include cystic hygroma, increased nuchal translucency, pleural effusion, and edema.[19]
Differential diagnosis
While Turner syndrome has similarities with renal anomalies and developmental delay, Turner syndrome is only found in females and often expresses differently. In Turner syndrome, there is a lower incidence of developmental delays, left-sided heart defects are constant and the occurrence of renal abnormalities is much lower.[20]
Other RASopathies
- Watson syndrome - Watson Syndrome has a number of similar characteristics with Noonan's Syndrome such as short stature, pulmonary valve stenosis, variable intellectual development, and skin pigment changes.[20][21]
- Cardiofaciocutaneous (CFC) syndrome - CFC syndrome is very similar to Noonan's Syndrome due to similar cardiac and lymphatic features. However, In CFC syndrome intellectual disability and gastrointestinal problems are often more severe and pronounced.[20][22]
- Costello syndrome - Like CFC syndrome, Costello syndrome has overlapping features with Noonan's Syndrome. However, the conditions can be distinguished by their genetic cause.[23][24]
- Neurofibromatosis 1 (NF1) [20][25]
- Williams syndrome[20][26]
Management
The treatment varies depending on complications but tend to be quite standard, reflecting the treatment of the general population.[20] Management guidelines, divided by systems, including general, developmental, dental, growth and feeding, cardiovascular, audiological, haematological, renal and skeletal, that account for actions to be taken at diagnosis, after diagnosis and if symptomatic, have been published by an American consortium.[19]
Specifically, treatment of cardiovascular complications resemble that of the general population and treatment of bleeding diathesis is guided by the specific factor deficiency or platelet aggregation.[20]
- Neuropsychological testing is recommended to find strengths and challenges to tailor support needed for school and career.
- Educational customization such as an individualized education program plan is sometimes needed for school-aged children.
- Speech therapy if speech and articulation issues present
- Physical therapy and occupational therapy for gross- and fine-motor delays
- Hypotonia and motor difficulties often impact handwriting. Accommodations for lessening handwriting demands will improve performance and save long-term hand function.
- Periodic follow up and lifelong monitoring of abnormalities found in any system, especially the cardiovascular system, is recommended.[27][8]
Anesthesia risk
Although a few people with Noonan syndrome have been reported to develop malignant hyperthermia, the gene mutation of diseases known to be associated with malignant hyperthermia is different from that of Noonan syndrome.[28]
Prognosis
The lifespan of people with Noonan's syndrome can be similar to the general population, however, Noonan syndrome can be associated with several health conditions that can contribute to mortality. The greatest contributor to mortality in individuals with Noonan syndrome is complications of cardiovascular disease.[29][8] Prognosis is therefore largely dependent on the presence or absence of cardiac disease, as well as the type and severity of the disease (if disease is present).[8] Most notably, Noonan syndrome with hypertrophic cardiomyopathy is associated with increased mortality.[29][8]
History

Jacqueline Noonan was practicing as a pediatric cardiologist at the University of Iowa when she noticed that children with a rare type of heart defect, valvular pulmonary stenosis, often had a characteristic physical appearance, with short stature, webbed neck, wide spaced eyes, and low-set ears. Both boys and girls were affected. These characteristics were sometimes seen running in families but were not associated with gross chromosomal abnormalities. She studied 833 people with Noonan syndrome at the congenital heart disease clinic, looking for other congenital abnormalities, and, in 1963, presented a paper: "Associated non-cardiac malformations in children with congenital heart disease".[30] This described nine children who in addition to congenital heart disease had characteristic facial features, chest deformities and short stature.
Dr. John Opitz, a former student of Noonan's, first began to call the condition "Noonan syndrome" when he saw children who looked like those whom Dr. Noonan had described. Noonan produced a paper titled "Hypertelorism with Turner Phenotype" in 1968 where she studied 19 patients who displayed symptoms indicative of Noonan's Syndrome.[31] In 1971, at the Symposium of Cardiovascular defects, the name "Noonan syndrome" became officially recognized.
References
- "Noonan syndrome". Genetics Home Reference. Retrieved 24 December 2018.
- "Noonan Syndrome". NORD (National Organization for Rare Disorders). 2016. Retrieved 24 December 2018.
- "Noonan syndrome". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Retrieved 25 December 2018.
- Bhambhani V, Muenke M (January 2014). "Noonan syndrome". American Family Physician. 89 (1): 37–43. PMC 4099190. PMID 24444506.
- "Noonan Syndrome - Children's Health Issues". Merck Manuals Consumer Version. Retrieved 25 December 2018.
- Nosan G, Bertok S, Vesel S, Yntema HG, Paro-Panjan D (December 2013). "A lethal course of hypertrophic cardiomyopathy in Noonan syndrome due to a novel germline mutation in the KRAS gene: case study". Croatian Medical Journal. 54 (6): 574–8. doi:10.3325/cmj.2013.54.574. PMC 3893993. PMID 24382853.
- Romano, Alicia A.; Allanson, Judith E.; Dahlgren, Jovanna; Gelb, Bruce D.; Hall, Bryan; Pierpont, Mary Ella; Roberts, Amy E.; Robinson, Wanda; Takemoto, Clifford M.; Noonan, Jacqueline A. (October 2010). "Noonan syndrome: clinical features, diagnosis, and management guidelines". Pediatrics. 126 (4): 746–759. doi:10.1542/peds.2009-3207. ISSN 1098-4275. PMID 20876176. S2CID 11297756.
- Allanson, Judith E.; Roberts, Amy E. (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Noonan Syndrome", GeneReviews®, University of Washington, Seattle, PMID 20301303, retrieved 2019-11-18
- Razzaque MA, Komoike Y, Nishizawa T, Inai K, Furutani M, Higashinakagawa T, Matsuoka R (March 2012). "Characterization of a novel KRAS mutation identified in Noonan syndrome". American Journal of Medical Genetics. Part A. 158A (3): 524–32. doi:10.1002/ajmg.a.34419. PMID 22302539. S2CID 34135931.
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. (December 2001). "Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome". Nature Genetics. 29 (4): 465–8. doi:10.1038/ng772. PMID 11704759. S2CID 14627986.
- Shchelochkov OA, Patel A, Weissenberger GM, Chinault AC, Wiszniewska J, Fernandes PH, et al. (April 2008). "Duplication of chromosome band 12q24.11q24.23 results in apparent Noonan syndrome". American Journal of Medical Genetics. Part A. 146A (8): 1042–8. doi:10.1002/ajmg.a.32215. PMID 18348260. S2CID 205309115.
- van Der Burgt I, Brunner H (September 2000). "Genetic heterogeneity in Noonan syndrome: evidence for an autosomal recessive form". American Journal of Medical Genetics. 94 (1): 46–51. doi:10.1002/1096-8628(20000904)94:1<46::AID-AJMG10>3.0.CO;2-I. PMID 10982482.
- Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, et al. (March 2006). "Germline KRAS mutations cause Noonan syndrome". Nature Genetics. 38 (3): 331–6. doi:10.1038/ng1748. PMID 16474405. S2CID 8193354.
- Bentires-Alj M, Kontaridis MI, Neel BG (March 2006). "Stops along the RAS pathway in human genetic disease". Nature Medicine. 12 (3): 283–5. doi:10.1038/nm0306-283. PMID 16520774. S2CID 6989331.
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, et al. (January 2007). "Germline gain-of-function mutations in SOS1 cause Noonan syndrome". Nature Genetics. 39 (1): 70–4. doi:10.1038/ng1926. PMID 17143285. S2CID 10222262.
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, et al. (August 2007). "Germline gain-of-function mutations in RAF1 cause Noonan syndrome". Nature Genetics. 39 (8): 1013–7. doi:10.1038/ng2078. PMID 17603482. S2CID 29753972.
- "Download Catalog - Mayo Medical Laboratories". www.mayomedicallaboratories.com. Archived from the original on 2017-05-17. Retrieved 2014-07-08.
- De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, et al. (December 2005). "NF1 gene mutations represent the major molecular event underlying neurofibromatosis–Noonan syndrome". American Journal of Human Genetics. 77 (6): 1092–101. doi:10.1086/498454. PMC 1285166. PMID 16380919.
- Roberts, Amy E.; Allanson, Judith E.; Tartaglia, Marco; Gelb, Bruce D. (2013-01-26). "Noonan syndrome". Lancet. 381 (9863): 333–342. doi:10.1016/S0140-6736(12)61023-X. ISSN 1474-547X. PMC 4267483. PMID 23312968.
- Allanson, Judith E.; Roberts, Amy E. (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Noonan Syndrome", GeneReviews®, University of Washington, Seattle, PMID 20301303, retrieved 2019-11-25
- Allanson, J. E.; Upadhyaya, M.; Watson, G. H.; Partington, M.; MacKenzie, A.; Lahey, D.; MacLeod, H.; Sarfarazi, M.; Broadhead, W.; Harper, P. S. (November 1991). "Watson syndrome: is it a subtype of type 1 neurofibromatosis?". Journal of Medical Genetics. 28 (11): 752–756. doi:10.1136/jmg.28.11.752. ISSN 0022-2593. PMC 1017110. PMID 1770531.
- Armour, C. M.; Allanson, J. E. (April 2008). "Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations". Journal of Medical Genetics. 45 (4): 249–254. doi:10.1136/jmg.2007.054460. ISSN 1468-6244. PMID 18039946. S2CID 9742395.
- Kerr, B.; Delrue, M.-A.; Sigaudy, S.; Perveen, R.; Marche, M.; Burgelin, I.; Stef, M.; Tang, B.; Eden, O. B.; O'Sullivan, J.; De Sandre-Giovannoli, A. (May 2006). "Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases". Journal of Medical Genetics. 43 (5): 401–405. doi:10.1136/jmg.2005.040352. ISSN 1468-6244. PMC 2564514. PMID 16443854.
- Gripp, Karen W.; Lin, Angela E.; Stabley, Deborah L.; Nicholson, Linda; Scott, Charles I.; Doyle, Daniel; Aoki, Yoko; Matsubara, Yoichi; Zackai, Elaine H.; Lapunzina, Pablo; Gonzalez-Meneses, Antonio (2006-01-01). "HRAS mutation analysis in Costello syndrome: Genotype and phenotype correlation". American Journal of Medical Genetics Part A. 140A (1): 1–7. doi:10.1002/ajmg.a.31047. ISSN 1552-4825. PMID 16329078. S2CID 27334655.
- Bertola, Debora R.; Pereira, Alexandre C.; Passetti, Fábio; de Oliveira, Paulo S.L.; Messiaen, Ludwine; Gelb, Bruce D.; Kim, Chong A.; Krieger, José Eduardo (2005-07-30). "Neurofibromatosis–Noonan syndrome: Molecular evidence of the concurrence of both disorders in a patient". American Journal of Medical Genetics Part A. 136A (3): 242–245. doi:10.1002/ajmg.a.30813. ISSN 1552-4825. PMID 15948193. S2CID 40235559.
- Morris, Colleen A. (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Williams Syndrome", GeneReviews®, University of Washington, Seattle, PMID 20301427, retrieved 2019-11-25
- "Noonan syndrome - Symptoms, diagnosis and treatment | BMJ Best Practice". bestpractice.bmj.com. Retrieved 2019-11-18.
- "Does Noonan Syndrome Increase Malignant Hyperthermia Susceptibility? - MHAUS". www.mhaus.org. Retrieved 2019-11-25.
- "DynaMed". www.dynamed.com. Retrieved 2019-11-11.
- Noonan JA, Ehmke DA (1963). "Associated noncardiac malformations in children with congenital heart disease". Midwest Soc Pediatr Res. 63: 468–70.
- Noonan, Jacqueline A. (1968-10-01). "Hypertelorism With Turner Phenotype: A New Syndrome With Associated Congenital Heart Disease". American Journal of Diseases of Children. 116 (4): 373–80. doi:10.1001/archpedi.1968.02100020377005. ISSN 0002-922X. PMID 4386970.