OXCT1
3-oxoacid CoA-transferase 1 (OXCT1) is an enzyme that in humans is encoded by the OXCT1 gene.[5][6] It is also known as succinyl-CoA-3-oxaloacid CoA transferase (SCOT). Mutations in the OXCT1 gene are associated with succinyl-CoA:3-oxoacid CoA transferase deficiency.[7] This gene encodes a member of the 3-oxoacid CoA-transferase gene family. The encoded protein is a homodimeric mitochondrial matrix enzyme that plays a central role in extrahepatic ketone body catabolism by catalyzing the reversible transfer of coenzyme A (CoA) from succinyl-CoA to acetoacetate.[6]
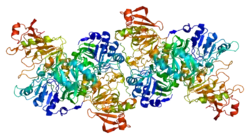
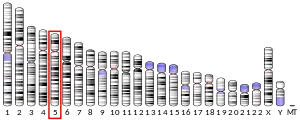


Structure
Gene
The OXCT1 gene resides on chromosome 5 at the band 5p13. OXCT1 spans a length of over 100 kb and includes 17 exons.[8]
Protein
The crystal structure of human OXCT1 reveals it to be a homodimer with two active sites. Each of its monomers contains N- and C-terminal domains that share an α/β structural fold characteristic of CoA transferase family I members. These terminal domains are joined by a linker region and form the enzyme's active site. Specifically, the conserved residue Glu344 within the active site is responsible for the enzyme's catalytic function by attacking the succinyl-CoA substrate, leading to the formation of the enzyme-CoA thioester intermediate.[9]
Function
OXCT1 is a member of the CoA transferase family I, which is known to catalyze the transfer of CoA between carboxylic acid groups.[9][10] In particular, OXCT1 catalyzes the first, rate-limiting step in ketolysis by transferring the CoA from succinyl-CoA to acetoacetate, giving acetoacetyl-CoA (AcAc-CoA). The product AcAc-CoA can then be converted by acetoacetyl-CoA thiolase into acetyl-CoA, which enters the citric acid cycle to generate energy for the cell.[9] As a result, OXCT1 allows cells to utilize energy stored in ketone bodies synthesized by the liver during conditions of energy deficiency, such as low glucose levels.[11] In addition, OXCT1 activity leads to the formation of Acetyl-CoA, which serves as a precursor for short-chain acyl-CoAs and lipids in the cytosol.[12]
OXCT1 is found in the mitochondrial matrix of all tissues except the liver, though it is most abundantly expressed in heart, brain, and kidney tissue.[9][11] Considering that liver cells function in ketogenesis and OXCT1 in ketolysis, OXCT1 may be absent from the liver to allow ketone body formation to proceed.[11]
Clinical Significance
Metabolic disorders
SCOT deficiency is a rare autosomal recessive metabolic disorder that can lead to recurrent episodes of ketoacidosis and even permanent ketosis. Twenty-four mutations in the human OXCT1 gene have been identified and associated with SCOT deficiency: three nonsense mutations, two insertion mutations, and 19 missense mutations. These mutations alter OXCT1 form and thus function in various ways, and they determine what phenotypic complications a patient may present. For instance, several missense mutations that substitute bulkier or charged residues hinder proper folding of OXCT1, leading to more severe outcomes such as permanent acidosis.[9]
OXCT1 has also been implicated diabetes. In a study by MacDonald et al., OXCT1 activity was shown to be lower by 92% in pancreatic islets of human patients with type 2 diabetes compared to those in healthy patients, though the cause is currently unknown.[12]
Cancer
Since OXCT1 functions in metabolizing ketone bodies, it has been proposed to promote tumor growth by providing tumor cells with an additional energy source. Therefore, ketone inhibitors may prove effective in treating patients experiencing recurring and metastatic tumors.[13] A proteomics study identified OXCT1 to be one of 16 proteins upregulated in carcinoma HepG2 cells treated with Platycodin D, an anti-cancer agent.[14]
References
- GRCh38: Ensembl release 89: ENSG00000083720 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000022186 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Kassovska-Bratinova S, Fukao T, Song XQ, Duncan AM, Chen HS, Robert MF, Pérez-Cerdá C, Ugarte M, Chartrand C, Vobecky S, Kondo N, Mitchell GA (September 1996). "Succinyl CoA: 3-oxoacid CoA transferase (SCOT): human cDNA cloning, human chromosomal mapping to 5p13, and mutation detection in a SCOT-deficient patient". American Journal of Human Genetics. 59 (3): 519–28. PMC 1914926. PMID 8751852.
- "Entrez Gene: OXCT1 3-oxoacid CoA transferase 1".
- Fukao T, Mitchell G, Sass JO, Hori T, Orii K, Aoyama Y (July 2014). "Ketone body metabolism and its defects". Journal of Inherited Metabolic Disease. 37 (4): 541–51. doi:10.1007/s10545-014-9704-9. PMID 24706027. S2CID 21840932.
- Fukao T, Mitchell GA, Song XQ, Nakamura H, Kassovska-Bratinova S, Orii KE, Wraith JE, Besley G, Wanders RJ, Niezen-Koning KE, Berry GT, Palmieri M, Kondo N (September 2000). "Succinyl-CoA:3-ketoacid CoA transferase (SCOT): cloning of the human SCOT gene, tertiary structural modeling of the human SCOT monomer, and characterization of three pathogenic mutations". Genomics. 68 (2): 144–51. doi:10.1006/geno.2000.6282. PMID 10964512.
- Shafqat N, Kavanagh KL, Sass JO, Christensen E, Fukao T, Lee WH, Oppermann U, Yue WW (November 2013). "A structural mapping of mutations causing succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency". Journal of Inherited Metabolic Disease. 36 (6): 983–7. doi:10.1007/s10545-013-9589-z. PMC 3825524. PMID 23420214.
- EMBL-EBI, InterPro. "Coenzyme A transferase family I (IPR004165) < InterPro < EMBL-EBI". www.ebi.ac.uk. Retrieved 2016-07-22.
- Orii KE, Fukao T, Song XQ, Mitchell GA, Kondo N (July 2008). "Liver-specific silencing of the human gene encoding succinyl-CoA: 3-ketoacid CoA transferase". The Tohoku Journal of Experimental Medicine. 215 (3): 227–36. doi:10.1620/tjem.215.227. PMID 18648183.
- MacDonald MJ, Longacre MJ, Langberg EC, Tibell A, Kendrick MA, Fukao T, Ostenson CG (June 2009). "Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes". Diabetologia. 52 (6): 1087–91. doi:10.1007/s00125-009-1319-6. PMC 2903059. PMID 19296078.
- Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP (November 2012). "Ketone body utilization drives tumor growth and metastasis". Cell Cycle. 11 (21): 3964–71. doi:10.4161/cc.22137. PMC 3507492. PMID 23082722.
- Lu JJ, Lu DZ, Chen YF, Dong YT, Zhang JR, Li T, Tang ZH, Yang Z (September 2015). "Proteomic analysis of hepatocellular carcinoma HepG2 cells treated with platycodin D". Chinese Journal of Natural Medicines. 13 (9): 673–9. doi:10.1016/S1875-5364(15)30065-0. PMID 26412427.
Further reading
- Pérez-Cerdá C, Merinero B, Sanz P, Jiménez A, Hernández C, García MJ, Ugarte M (1992). "A new case of succinyl-CoA: acetoacetate transferase deficiency". Journal of Inherited Metabolic Disease. 15 (3): 371–3. doi:10.1007/BF02435979. PMID 1405472. S2CID 13058612.
- Zołnierowicz S, Scisłowski PW, Swierczyński J, Zelewski L (1985). "Acetoacetate utilization by human placental mitochondria". Placenta. 5 (3): 271–6. doi:10.1016/S0143-4004(84)80037-5. PMID 6150478.
- Fukao T, Song XQ, Mitchell GA, Yamaguchi S, Sukegawa K, Orii T, Kondo N (October 1997). "Enzymes of ketone body utilization in human tissues: protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases". Pediatric Research. 42 (4): 498–502. doi:10.1203/00006450-199710000-00013. PMID 9380443.
- Song XQ, Fukao T, Watanabe H, Shintaku H, Hirayama K, Kassovska-Bratinova S, Kondo N, Mitchell GA (1998). "Succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency: two pathogenic mutations, V133E and C456F, in Japanese siblings". Human Mutation. 12 (2): 83–8. doi:10.1002/(SICI)1098-1004(1998)12:2<83::AID-HUMU2>3.0.CO;2-P. PMID 9671268. S2CID 42111273.
- Tanaka H, Kohroki J, Iguchi N, Onishi M, Nishimune Y (January 2002). "Cloning and characterization of a human orthologue of testis-specific succinyl CoA: 3-oxo acid CoA transferase (Scot-t) cDNA". Molecular Human Reproduction. 8 (1): 16–23. doi:10.1093/molehr/8.1.16. PMID 11756565.
- Fukao T, Shintaku H, Kusubae R, Zhang GX, Nakamura K, Kondo M, Kondo N (December 2004). "Patients homozygous for the T435N mutation of succinyl-CoA:3-ketoacid CoA Transferase (SCOT) do not show permanent ketosis". Pediatric Research. 56 (6): 858–63. doi:10.1203/01.PDR.0000145297.90577.67. PMID 15496607.
- Fukao T, Sakurai S, Rolland MO, Zabot MT, Schulze A, Yamada K, Kondo N (November 2006). "A 6-bp deletion at the splice donor site of the first intron resulted in aberrant splicing using a cryptic splice site within exon 1 in a patient with succinyl-CoA: 3-Ketoacid CoA transferase (SCOT) deficiency". Molecular Genetics and Metabolism. 89 (3): 280–2. doi:10.1016/j.ymgme.2006.04.014. PMID 16765626.
PDB gallery | |
|---|---|
|