Quantitative trait locus
A quantitative trait locus (QTL) is a locus (section of DNA) that correlates with variation of a quantitative trait in the phenotype of a population of organisms.[1] QTLs are mapped by identifying which molecular markers (such as SNPs or AFLPs) correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
Definition
A quantitative trait locus (QTL) is a region of DNA which is associated with a particular phenotypic trait, which varies in degree and which can be attributed to polygenic effects, i.e., the product of two or more genes, and their environment.[2] These QTLs are often found on different chromosomes. The number of QTLs which explain variation in the phenotypic trait indicates the genetic architecture of a trait. It may indicate that plant height is controlled by many genes of small effect, or by a few genes of large effect.
Typically, QTLs underlie continuous traits (those traits which vary continuously, e.g. height) as opposed to discrete traits (traits that have two or several character values, e.g. red hair in humans, a recessive trait, or smooth vs. wrinkled peas used by Mendel in his experiments).
Moreover, a single phenotypic trait is usually determined by many genes. Consequently, many QTLs are associated with a single trait. Another use of QTLs is to identify candidate genes underlying a trait. The DNA sequence of any genes in this region can then be compared to a database of DNA for genes whose function is already known, this task being fundamental for marker-assisted crop improvement.[3][4]
History
Mendelian inheritance was rediscovered at the beginning of the 20th century. As Mendel's ideas spread, geneticists began to connect Mendel's rules of inheritance of single factors to Darwinian evolution. For early geneticists, it was not immediately clear that the smooth variation in traits like body size (i.e., incomplete dominance) was caused by the inheritance of single genetic factors. Although Darwin himself observed that inbred features of fancy pigeons were inherited in accordance with Mendel's laws (although Darwin didn't actually know about Mendel's ideas when he made the observation), it was not obvious that these features selected by fancy pigeon breeders can similarly explain quantitative variation in nature.[5]
An early attempt by William Ernest Castle to unify the laws of Mendelian inheritance with Darwin's theory of speciation invoked the idea that species become distinct from one another as one species or the other acquires a novel Mendelian factor.[6] Castle's conclusion was based on the observation that novel traits that could be studied in the lab and that show Mendelian inheritance patterns reflect a large deviation from the wild type, and Castle believed that acquisition of such features is the basis of "discontinuous variation" that characterizes speciation.[6] Darwin discussed the inheritance of similar mutant features but did not invoke them as a requirement of speciation.[5] Instead Darwin used the emergence of such features in breeding populations as evidence that mutation can occur at random within breeding populations, which is a central premise of his model of selection in nature.[5] Later in his career, Castle would refine his model for speciation to allow for small variation to contribute to speciation over time. He also was able to demonstrate this point by selectively breeding laboratory populations of rats to obtain a hooded phenotype over several generations.[7]
Castle's was perhaps the first attempt made in the scientific literature to direct evolution by artificial selection of a trait with continuous underlying variation, however the practice had previously been widely employed in the development of agriculture to obtain livestock or plants with favorable features from populations that show quantitative variation in traits like body size or grain yield.
Castle's work was among the first to attempt to unify the recently rediscovered laws of Mendelian inheritance with Darwin's theory of evolution. Still, it would be almost thirty years until the theoretical framework for evolution of complex traits would be widely formalized.[8] In an early summary of the theory of evolution of continuous variation, Sewall Wright, a graduate student who trained under Castle, summarized contemporary thinking about the genetic basis of quantitative natural variation: "As genetic studies continued, ever smaller differences were found to mendelize, and any character, sufficiently investigated, turned out to be affected by many factors."[8] Wright and others formalized population genetics theory that had been worked out over the preceding 30 years explaining how such traits can be inherited and create stably breeding populations with unique characteristics. Quantitative trait genetics today leverages Wright's observations about the statistical relationship between genotype and phenotype in families and populations to understand how certain genetic features can affect variation in natural and derived populations.
Quantitative traits
Polygenic inheritance refers to inheritance of a phenotypic characteristic (trait) that is attributable to two or more genes and can be measured quantitatively. Multifactorial inheritance refers to polygenic inheritance that also includes interactions with the environment. Unlike monogenic traits, polygenic traits do not follow patterns of Mendelian inheritance (discrete categories). Instead, their phenotypes typically vary along a continuous gradient depicted by a bell curve.[9]
An example of a polygenic trait is human skin color variation. Several genes factor into determining a person's natural skin color, so modifying only one of those genes can change skin color slightly or in some cases, such as for SLC24A5, moderately. Many disorders with genetic components are polygenic, including autism, cancer, diabetes and numerous others. Most phenotypic characteristics are the result of the interaction of multiple genes.
Examples of disease processes generally considered to be results of many contributing factors:
Congenital malformation
- Cleft palate[10][11]
- Congenital dislocation of the hip[12]
- Congenital heart defects
- Neural tube defects
- Pyloric stenosis
- Talipes
Adult onset diseases
- Diabetes Mellitus
- Cancer[11]
- Glaucoma
- Hypertension
- Ischaemic heart disease
- Bipolar disorder
- Schizophrenia
- Psoriasis
- Thyroid diseases
- Alzheimer's disease
Multifactorially inherited diseases are said to constitute the majority of genetic disorders affecting humans which will result in hospitalization or special care of some kind.[13][14]
Multifactorial traits in general
Traits controlled both by the environment and by genetic factors are called multifactorial. Usually, multifactorial traits outside of illness result in what we see as continuous characteristics in organisms, especially human organisms such as: height,[13] skin color, and body mass.[15] All of these phenotypes are complicated by a great deal of give-and-take between genes and environmental effects.[13] The continuous distribution of traits such as height and skin color described above, reflects the action of genes that do not manifest typical patterns of dominance and recessiveness. Instead the contributions of each involved locus are thought to be additive. Writers have distinguished this kind of inheritance as polygenic, or quantitative inheritance.[16]
Thus, due to the nature of polygenic traits, inheritance will not follow the same pattern as a simple monohybrid or dihybrid cross.[14] Polygenic inheritance can be explained as Mendelian inheritance at many loci,[13] resulting in a trait which is normally-distributed. If n is the number of involved loci, then the coefficients of the binomial expansion of (a + b)2n will give the frequency of distribution of all n allele combinations. For sufficiently high values of n, this binomial distribution will begin to resemble a normal distribution. From this viewpoint, a disease state will become apparent at one of the tails of the distribution, past some threshold value. Disease states of increasing severity will be expected the further one goes past the threshold and away from the mean.[16]
Heritable disease and multifactorial inheritance
A mutation resulting in a disease state is often recessive, so both alleles must be mutant in order for the disease to be expressed phenotypically. A disease or syndrome may also be the result of the expression of mutant alleles at more than one locus. When more than one gene is involved, with or without the presence of environmental triggers, we say that the disease is the result of multifactorial inheritance.
The more genes involved in the cross, the more the distribution of the genotypes will resemble a normal, or Gaussian distribution.[13] This shows that multifactorial inheritance is polygenic, and genetic frequencies can be predicted by way of a polyhybrid Mendelian cross. Phenotypic frequencies are a different matter, especially if they are complicated by environmental factors.
The paradigm of polygenic inheritance as being used to define multifactorial disease has encountered much disagreement. Turnpenny (2004) discusses how simple polygenic inheritance cannot explain some diseases such as the onset of Type I diabetes mellitus, and that in cases such as these, not all genes are thought to make an equal contribution.[16]
The assumption of polygenic inheritance is that all involved loci make an equal contribution to the symptoms of the disease. This should result in a normal (Gaussian) distribution of genotypes. When it does not, the idea of polygenetic inheritance cannot be supported for that illness.
Examples
The above are well-known examples of diseases having both genetic and environmental components. Other examples involve atopic diseases such as eczema or dermatitis;[13]spina bifida (open spine), and anencephaly (open skull).[10]
While schizophrenia is widely believed to be multifactorially genetic by biopsychiatrists, no characteristic genetic markers have been determined with any certainty.
If it is shown that the brothers and sisters of the patient have the disease, then there is a strong chance that the disease is genetic and that the patient will also be a genetic carrier. This is not quite enough as it also needs to be proven that the pattern of inheritance is non-Mendelian. This would require studying dozens, even hundreds of different family pedigrees before a conclusion of multifactorial inheritance is drawn. This often takes several years.
If multifactorial inheritance is indeed the case, then the chance of the patient contracting the disease is reduced only if cousins and more distant relatives have the disease.[10] It must be stated that while multifactorially-inherited diseases tend to run in families, inheritance will not follow the same pattern as a simple monohybrid or dihybrid cross.[14]
If a genetic cause is suspected and little else is known about the illness, then it remains to be seen exactly how many genes are involved in the phenotypic expression of the disease. Once that is determined, the question must be answered: if two people have the required genes, why are there differences in expression between them? Generally, what makes the two individuals different are likely to be environmental factors. Due to the involved nature of genetic investigations needed to determine such inheritance patterns, this is not usually the first avenue of investigation one would choose to determine etiology.
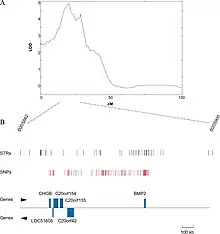
QTL mapping

For organisms whose genomes are known, one might now try to exclude genes in the identified region whose function is known with some certainty not to be connected with the trait in question. If the genome is not available, it may be an option to sequence the identified region and determine the putative functions of genes by their similarity to genes with known function, usually in other genomes. This can be done using BLAST, an online tool that allows users to enter a primary sequence and search for similar sequences within the BLAST database of genes from various organisms. It is often not the actual gene underlying the phenotypic trait, but rather a region of DNA that is closely linked with the gene[17][18]
Another interest of statistical geneticists using QTL mapping is to determine the complexity of the genetic architecture underlying a phenotypic trait. For example, they may be interested in knowing whether a phenotype is shaped by many independent loci, or by a few loci, and do those loci interact. This can provide information on how the phenotype may be evolving.[19]
In a recent development, classical QTL analyses were combined with gene expression profiling i.e. by DNA microarrays. Such expression QTLs (eQTLs) describe cis- and trans-controlling elements for the expression of often disease-associated genes.[20] Observed epistatic effects have been found beneficial to identify the gene responsible by a cross-validation of genes within the interacting loci with metabolic pathway- and scientific literature databases.
Analysis of variance
The simplest method for QTL mapping is analysis of variance (ANOVA, sometimes called "marker regression") at the marker loci. In this method, in a backcross, one may calculate a t-statistic to compare the averages of the two marker genotype groups. For other types of crosses (such as the intercross), where there are more than two possible genotypes, one uses a more general form of ANOVA, which provides a so-called F-statistic. The ANOVA approach for QTL mapping has three important weaknesses. First, we do not receive separate estimates of QTL location and QTL effect. QTL location is indicated only by looking at which markers give the greatest differences between genotype group averages, and the apparent QTL effect at a marker will be smaller than the true QTL effect as a result of recombination between the marker and the QTL. Second, we must discard individuals whose genotypes are missing at the marker. Third, when the markers are widely spaced, the QTL may be quite far from all markers, and so the power for QTL detection will decrease.
Interval mapping
Lander and Botstein developed interval mapping, which overcomes the three disadvantages of analysis of variance at marker loci.[21] Interval mapping is currently the most popular approach for QTL mapping in experimental crosses. The method makes use of a genetic map of the typed markers, and, like analysis of variance, assumes the presence of a single QTL. In interval mapping, each locus is considered one at a time and the logarithm of the odds ratio (LOD score) is calculated for the model that the given locus is a true QTL. The odds ratio is related to the Pearson correlation coefficient between the phenotype and the marker genotype for each individual in the experimental cross.[22]
The term 'interval mapping' is used for estimating the position of a QTL within two markers (often indicated as 'marker-bracket'). Interval mapping is originally based on the maximum likelihood but there are also very good approximations possible with simple regression.
The principle for QTL mapping is: 1) The likelihood can be calculated for a given set of parameters (particularly QTL effect and QTL position) given the observed data on phenotypes and marker genotypes. 2) The estimates for the parameters are those where the likelihood is highest. 3) A significance threshold can be established by permutation testing.[23]
Conventional methods for the detection of quantitative trait loci (QTLs) are based on a comparison of single QTL models with a model assuming no QTL. For instance in the "interval mapping" method[24] the likelihood for a single putative QTL is assessed at each location on the genome. However, QTLs located elsewhere on the genome can have an interfering effect. As a consequence, the power of detection may be compromised, and the estimates of locations and effects of QTLs may be biased (Lander and Botstein 1989; Knapp 1991). Even nonexisting so-called "ghost" QTLs may appear (Haley and Knott 1992; Martinez and Curnow 1992). Therefore, multiple QTLs could be mapped more efficiently and more accurately by using multiple QTL models.[25] One popular approach to handle QTL mapping where multiple QTL contribute to a trait is to iteratively scan the genome and add known QTL to the regression model as QTLs are identified. This method, termed composite interval mapping determine both the location and effects size of QTL more accurately than single-QTL approaches, especially in small mapping populations where the effect of correlation between genotypes in the mapping population may be problematic.
Composite interval mapping (CIM)
In this method, one performs interval mapping using a subset of marker loci as covariates. These markers serve as proxies for other QTLs to increase the resolution of interval mapping, by accounting for linked QTLs and reducing the residual variation. The key problem with CIM concerns the choice of suitable marker loci to serve as covariates; once these have been chosen, CIM turns the model selection problem into a single-dimensional scan. The choice of marker covariates has not been solved, however. Not surprisingly, the appropriate markers are those closest to the true QTLs, and so if one could find these, the QTL mapping problem would be complete anyway.
Inclusive composite interval mapping (ICIM) has also been proposed as a potential method for QTL mapping.[26]
Family-pedigree based mapping
Family-based QTL mapping, or Family-pedigree based mapping (Linkage and association mapping), involves multiple families instead of a single family. Family-based QTL mapping has been the only way for mapping of genes where experimental crosses are difficult to make. However, due to some advantages, now plant geneticists are attempting to incorporate some of the methods pioneered in human genetics.[27] Using family-pedigree based approach has been discussed (Bink et al. 2008). Family-based linkage and association has been successfully implemented (Rosyara et al. 2009)[28]
See also
References
- Miles, C; Wayne, M (2008). "Quantitative trait locus (QTL) analysis". Nature Education. 1 (1).
- Complex Trait Consortium —; Abiola, Oduola; Angel, Joe M.; Avner, Philip; Bachmanov, Alexander A.; Belknap, John K.; Bennett, Beth; Blankenhorn, Elizabeth P.; Blizard, David A.; Bolivar, Valerie; Brockmann, Gudrun A.; Buck, Kari J.; Bureau, Jean-Francois; Casley, William L.; Chesler, Elissa J.; Cheverud, James M.; Churchill, Gary A.; Cook, Melloni; Crabbe, John C.; Crusio, Wim E.; Darvasi, Ariel; de Haan, Gerald; Demant, Peter; Doerge, R. W.; Elliott, Rosemary W.; Farber, Charles R.; Flaherty, Lorraine; Flint, Jonathan; Gershenfeld, Howard; Gibson, John P.; Gu, Jing; Gu, Weikuan; Himmelbauer, Heinz; Hitzemann, Robert; Hsu, Hui-Chen; Hunter, Kent; Iraqi, Fuad A.; Jansen, Ritsert C.; Johnson, Thomas E.; Jones, Byron C.; Kempermann, Gerd; Lammert, Frank; Lu, Lu; Manly, Kenneth F.; Matthews, Douglas B.; Medrano, Juan F.; Mehrabian, Margarete; Mittleman, Guy; Mock, Beverly A.; Mogil, Jeffrey S.; Montagutelli, Xavier; Morahan, Grant; Mountz, John D.; Nagase, Hiroki; Nowakowski, Richard S.; O’Hara, Bruce F.; Osadchuk, Alexander V.; Paigen, Beverly; Palmer, Abraham A.; Peirce, Jeremy L.; Pomp, Daniel; Rosemann, Michael; Rosen, Glenn D.; Schalkwyk, Leonard C.; Seltzer, Ze’ev; Settle, Stephen; Shimomura, Kazuhiro; Shou, Siming; Sikela, James M.; Siracusa, Linda D.; Spearow, Jimmy L.; Teuscher, Cory; Threadgill, David W.; Toth, Linda A.; Toye, Ayo A.; Vadasz, Csaba; Van Zant, Gary; Wakeland, Edward; Williams, Robert W.; Zhang, Huang-Ge; Zou, Fei (2003). "The nature and identification of quantitative trait loci: a community's view". Nature Reviews Genetics. Nature Portfolio. 4 (11): 911–916. doi:10.1038/nrg1206. PMC 2063446. PMID 14634638. S2CID 27285742. S2CID 195367115.
- Watanabe, Satoshi; Hideshima, Rumiko; Xia, Zhengjun; et al. (2009). "Map-Based Cloning of the Gene Associated With the Soybean Maturity Locus E3". Genetics. 182 (4): 1251–1262. doi:10.1534/genetics.108.098772. PMC 2728863. PMID 19474204.
- Daware, Anurag; Parida, Swarup K.; Tyagi, Akhilesh K. (2020), Vaschetto, Luis M. (ed.), "Integrated Genomic Strategies for Cereal Genetic Enhancement: Combining QTL and Association Mapping", Cereal Genomics: Methods and Protocols, Methods in Molecular Biology, Springer US, vol. 2072, pp. 15–25, doi:10.1007/978-1-4939-9865-4_3, ISBN 9781493998654, PMID 31541435, S2CID 202711099
- "Archived copy". Archived from the original on 3 October 2013. Retrieved 24 September 2013.
{{cite web}}: CS1 maint: archived copy as title (link) - Castle WE (1903). "Mendel's Law of Heredity". Science. 18 (456): 396–406. Bibcode:1903Sci....18..396C. doi:10.1126/science.18.456.396. PMID 17752783. S2CID 11670642.
- Castle, W. E. (1 May 1951). "Variation in the Hooded Pattern of Rats, and a New Allele of Hooded". Genetics. 36 (3): 254–266. doi:10.1093/genetics/36.3.254. PMC 1209518. PMID 14840647 – via www.genetics.org.
- Wright, Sewall (1 March 1931). "Evolution in Mendelian Populations". Genetics. 16 (2): 97–159. doi:10.1093/genetics/16.2.97. PMC 1201091. PMID 17246615 – via www.genetics.org.
- Ricki Lewis (2003), Multifactorial Traits, McGraw-Hill Higher Education.
- Proud, Virginia & Roberts, Helen (31 December 2005). "Medical Genetics: Multifactorial Inheritance". Children's Hospital of the King's Daughters. Archived from the original on 15 October 2006. Retrieved 6 January 2007.
- "Multifactorial Inheritance". Pregnancy and Newborn Health Education Centre. The March of Dimes. Archived from the original on 2 November 2006. Retrieved 12 November 2014.
- Emery's Elements of Medical Genetics
- Tissot, Robert. "Human Genetics for 1st Year Students: Multifactorial Inheritance". Retrieved 6 January 2007.
- Birth Defects Genetics Centre, University of South Dakota School of Medicine. "Multifactorial Inheritance". Clinical Genetics: A Self-Study Guide for Health Care Providers. University of South Dakota School of Medicine. Archived from the original on 30 December 2006. Retrieved 6 January 2007.
- "Definition of Multifactorial inheritance". MedicineNet.com MedTerms Dictionary. MedicineNet.com. Archived from the original on 17 December 2013. Retrieved 6 January 2007.
- Turnpenny, Peter (2004). "Chapter 9" (PDF). Emery's Elements of Medical Genetics (12th ed.). Elsevier. Retrieved 6 January 2007.
- "BLAST: Basic Local Alignment Search Tool". blast.ncbi.nlm.nih.gov. Retrieved 18 February 2018.
- Daware, Anurag; Parida, Swarup K.; Tyagi, Akhilesh K. (2020), Vaschetto, Luis M. (ed.), "Integrated Genomic Strategies for Cereal Genetic Enhancement: Combining QTL and Association Mapping", Cereal Genomics: Methods and Protocols, Methods in Molecular Biology, Springer US, vol. 2072, pp. 15–25, doi:10.1007/978-1-4939-9865-4_3, ISBN 9781493998654, PMID 31541435, S2CID 202711099
- Grisel, Judith E.; Crabbe, John C. (1995). "Quantitative Trait Loci Mapping". Alcohol Health and Research World. 19 (3): 220–227. ISSN 0090-838X. PMC 6875759. PMID 31798043.
- Westra HJ, et al. (2013). "Systematic identification of trans eQTLs as putative drivers of known disease associations". Nat Genet. 45 (10): 1238–1243. doi:10.1038/ng.2756. PMC 3991562. PMID 24013639.
- Lander, E.S.; Botstein, D. (1989). "Mapping mendelian factors underlying quantitative traits using RFLP linkage maps". Genetics. 121 (1): 185–199. doi:10.1093/genetics/121.1.185. PMC 1203601. PMID 2563713.
- Lynch, M. & Walsh, B. Genetics and Analysis of Quantitative Traits edn 1 (Sinauer Associates, 1998).
- Bloom J. S.; Ehrenreich I. M.; Loo W. T.; Lite T.-L. V.; Kruglyak L. (2013). "Finding the sources of missing heritability in a yeast cross". Nature. 494 (7436): 234–237. arXiv:1208.2865. Bibcode:2013Natur.494..234B. doi:10.1038/nature11867. PMC 4001867. PMID 23376951.
- Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. ES Lander and D Botstein. Genetics. 1989
- Jansen, R C (1 September 1993). "Interval mapping of multiple quantitative trait loci" (PDF). Genetics. 135 (1): 205–211. doi:10.1093/genetics/135.1.205. PMC 1205619. PMID 8224820. Retrieved 1 March 2023.
- Li, Shanshan; Wang, Jiankang; Zhang, Luyan (10 July 2015). "Inclusive Composite Interval Mapping of QTL by Environment Interactions in Biparental Populations". PLOS ONE. 10 (7): e0132414. Bibcode:2015PLoSO..1032414L. doi:10.1371/journal.pone.0132414. ISSN 1932-6203. PMC 4498613. PMID 26161656.
- Jannink, J; Bink, Mc; Jansen, Rc (August 2001). "Using complex plant pedigrees to map valuable genes". Trends in Plant Science. 6 (8): 337–42. doi:10.1016/S1360-1385(01)02017-9. ISSN 1360-1385. PMID 11495765.
- Rosyara, U. R.; Maxson-stein, K.L.; Glover, K.D.; Stein, J.M.; Gonzalez-hernandez, J.L. (2007). "Family-based mapping of FHB resistance QTLs in hexaploid wheat". Proceedings of National Fusarium Head Blight Forum.
- Bink MCAM, Boer MP, ter Braak CJF, Jansen J, Voorrips RE, van de Weg WE: Bayesian analysis of complex traits in pedigreed plant populations.
Euphytica 2008, 161:85–96.
- Rosyara U.R., J.L. Gonzalez-Hernandez, K.D. Glover, K.R. Gedye and J.M. Stein. 2009. Family-based mapping of quantitative trait loci in plant breeding populations with resistance to Fusarium head blight in wheat as an illustration Theoretical Applied Genetics 118:1617–1631
- Garnier, Sophie, Truong, Vinh, Genome-Wide Haplotype Analysis of Cis Expression Quantitative Trait Loci in Monocytes
External links
- Plant Breeding and Genomics on eXtension.org
- INTERSNP – a software for genome-wide interaction analysis (GWIA) of case-control SNP data and analysis of quantitative traits
- Precision Mapping of Quantitative Trait Loci
- QTL Cartographer
- Complex Trait Consortium
- A Statistical Framework for Quantitative Trait Mapping
- GeneNetwork
- GridQTL
- QTL discussion forum
- A list of computer programs for genetic analysis including QTL analysis
- Quantitative Trait Locus (QTL) Analysis @ Scitable
- Mapping Quantitative Trait Loci
- What are Quantitative Trait Loci? – University of Warwick