Protein aggregation
In molecular biology, protein aggregation is a phenomenon in which intrinsically-disordered or mis-folded proteins aggregate (i.e., accumulate and clump together) either intra- or extracellularly.[1][2] Protein aggregates have been implicated in a wide variety of diseases known as amyloidoses, including ALS, Alzheimer's, Parkinson's and prion disease.[3][4]
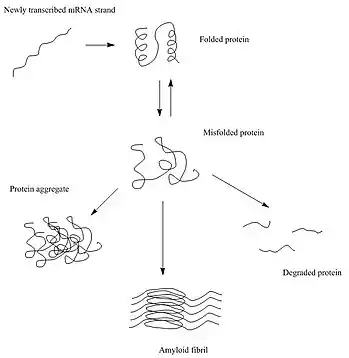
After synthesis, proteins typically fold into a particular three-dimensional conformation that is the most thermodynamically favorable: their native state.[5] This folding process is driven by the hydrophobic effect: a tendency for hydrophobic (water-fearing) portions of the protein to shield themselves from the hydrophilic (water-loving) environment of the cell by burying into the interior of the protein. Thus, the exterior of a protein is typically hydrophilic, whereas the interior is typically hydrophobic.
Protein structures are stabilized by non-covalent interactions and disulfide bonds between two cysteine residues. The non-covalent interactions include ionic interactions and weak van der Waals interactions. Ionic interactions form between an anion and a cation and form salt bridges that help stabilize the protein. Van der Waals interactions include nonpolar interactions (i.e. London dispersion force) and polar interactions (i.e. hydrogen bonds, dipole-dipole bond). These play an important role in a protein's secondary structure, such as forming an alpha helix or a beta sheet, and tertiary structure. Interactions between amino acid residues in a specific protein are very important in that protein's final structure.
When there are changes in the non-covalent interactions, as may happen with a change in the amino acid sequence, the protein is susceptible to misfolding or unfolding. In these cases, if the cell does not assist the protein in re-folding, or degrade the unfolded protein, the unfolded/misfolded protein may aggregate, in which the exposed hydrophobic portions of the protein may interact with the exposed hydrophobic patches of other proteins.[6][7] There are three main types of protein aggregates that may form: amorphous aggregates, oligomers, and amyloid fibrils.[8]
Causes
Protein aggregation can occur due to a variety of causes. There are four classes that these causes can be categorized into, which are detailed below.
Mutations
Mutations that occur in the DNA sequence may or may not affect the amino acid sequence of the protein. When the sequence is affected, a different amino acid may change the interactions between the side chains that affect the folding of the protein. This can lead to exposed hydrophobic regions of the protein that aggregate with the same misfolded/unfolded protein or a different protein.[9]
In addition to mutations in the affected proteins themselves, protein aggregation could also be caused indirectly through mutations in proteins in regulatory pathways such as the refolding pathway (molecular chaperones) or the ubiquitin-proteasome pathway (ubiquitin ligases).[10] Chaperones help with protein refolding by providing a safe environment for the protein to fold. Ubiquitin ligases target proteins for degradation through ubiquitin modification.[11]
Problems with protein synthesis
Protein aggregation can be caused by problems that occur during transcription or translation. During transcription, DNA is copied into mRNA, forming a strand of pre-mRNA that undergoes RNA processing to form mRNA.[12] During translation, ribosomes and tRNA help translate the mRNA sequence into an amino acid sequence.[12] If problems arise during either step, making an incorrect mRNA strand and/or an incorrect amino acid sequence, this can cause the protein to misfold, leading to protein aggregation.
Environmental stresses
Environmental stresses such as extreme temperatures and pH or oxidative stress can also lead to protein aggregation.[13] One such disease is cryoglobulinemia.
Extreme temperatures can weaken and destabilize the non-covalent interactions between the amino acid residues. pHs outside of the protein's pH range can change the protonation state of the amino acids, which can increase or decrease the non-covalent interactions. This can also lead to less stable interactions and result in protein unfolding.
Oxidative stress can be caused by radicals such as reactive oxygen species (ROS). These unstable radicals can attack the amino acid residues, leading to oxidation of side chains (e.g. aromatic side chains, methionine side chains) and/or cleavage of the polypeptide bonds.[14] This can affect the non-covalent interactions that hold the protein together correctly, which can cause protein destabilization, and may cause the protein to unfold.[13]
Aging
Cells have mechanisms that can refold or degrade protein aggregates. However, as cells age, these control mechanisms are weakened and the cell is less able to resolve the aggregates.[13]
The hypothesis that protein aggregation is a causative process in aging is testable now since some models of delayed aging are in hand. If the development of protein aggregates was an aging independent process, slowing down aging will show no effect on the rate of proteotoxicity over time. However, if aging is associated with decline in the activity of protective mechanisms against proteotoxicity, the slow aging models would show reduced aggregation and proteotoxicity. To address this problem several toxicity assays have been done in C. elegans. These studies indicated that reducing the activity of insulin/IGF signaling (IIS), a prominent aging regulatory pathway protects from neurodegeneration-linked toxic protein aggregation. The validity of this approach has been tested and confirmed in mammals as reducing the activity of the IGF-1 signaling pathway protected Alzheimer's model mice from the behavioral and biochemical impairments associated with the disease.[15]
Aggregate localization
Several studies have shown that cellular responses to protein aggregation are well-regulated and organized. Protein aggregates localize to specific areas in the cell, and research has been done on these localizations in prokaryotes (E.coli) and eukaryotes (yeast, mammalian cells).[16] From the macroscopic point of view, positron emission tomography tracers are used for certain misfolded proitein.[17] Recently, a team of researchers led by Dr. Alessandro Crimi has proposed a machine learning method to predict future deposition in the brain. [18]
Bacteria
The aggregates in bacteria asymmetrically end up at one of the poles of the cell, the "older pole." After the cell divides, the daughter cells with the older pole gets the protein aggregate and grows more slowly than daughter cells without the aggregate. This provides a natural selection mechanism for reducing protein aggregates in the bacterial population.[19]
Yeast
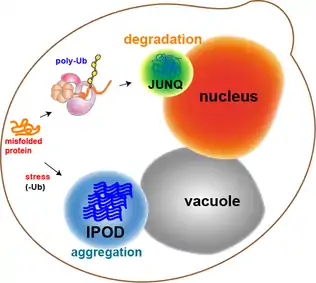
Most of the protein aggregates in yeast cells get refolded by molecular chaperones. However, some aggregates, such as the oxidatively damaged proteins or the proteins marked for degradation, cannot be refolded. Rather, there are two compartments that they can end up in. Protein aggregates can be localized at the Juxtanuclear quality-control compartment (JUNQ), which is near the nuclear membrane, or at the Insoluble Protein deposit (IPOD), near the vacuole in yeast cells.[13] Protein aggregates localize at JUNQ when they are ubiquitinated and targeted for degradation. The aggregated and insoluble proteins localize at IPOD as a more permanent deposition. There is evidence that the proteins here may be removed by autophagy.[20] These two pathways work together in that the proteins tend to come to the IPOD when the proteasome pathway is being overworked.[20]
Mammalian cells
In mammalian cells, these protein aggregates are termed "aggresomes" and they are formed when the cell is diseased. This is because aggregates tend to form when there are heterologous proteins present in the cell, which can arise when the cell is mutated. Different mutates of the same protein may form aggresomes of different morphologies, ranging from diffuse dispersion of soluble species to large puncta, which in turn bear different pathogenicity.[21] The E3 ubiquitin ligase is able to recognize misfolded proteins and ubiquinate them. HDAC6 can then bind to the ubiquitin and the motor protein dynein to bring the marked aggregates to the microtubule organizing center (MTOC). There, they pack together into a sphere that surrounds the MTOC. They bring over chaperones and proteasomes and activate autophagy.[22]
Elimination
There are two main protein quality control systems in the cell that are responsible for eliminating protein aggregates. Misfolded proteins can get refolded by the bi-chaperone system or degraded by the ubiquitin proteasome system or autophagy.[23]
Refolding
The bi-chaperone system utilizes the Hsp70 (DnaK-DnaJ-GrpE in E. coli and Ssa1-Ydj1/Sis1-Sse1/Fe1 in yeast) and Hsp100 (ClpB in E. coli and Hsp104 in yeast) chaperones for protein disaggregation and refolding.[24]
Hsp70 interacts with the protein aggregates and recruits Hsp100. Hsp70 stabilizes an activated Hsp100. Hsp100 proteins have aromatic pore loops that are used for threading activity to disentangle single polypeptides. This threading activity can be initiated at the N-terminus, C-terminus or in the middle of the polypeptide. The polypeptide gets translocated through Hsp100 in a series of steps, utilizing an ATP at each step.[24] The polypeptide unfolds and is then allowed to refold either by itself or with the help of heat shock proteins.[25]
Degradation
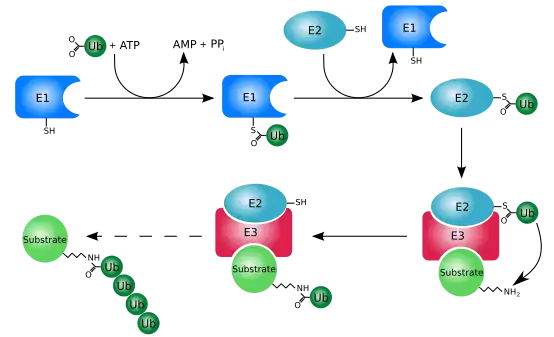
Misfolded proteins can be eliminated through the ubiquitin-proteasome system (UPS). This consists of an E1-E2-E3 pathway that ubiquinates proteins to mark them for degradation. In eukaryotes, the proteins get degraded by the 26S proteasome. In mammalian cells, the E3 ligase, carboxy-terminal Hsp70 interacting protein (CHIP), targets Hsp70-bound proteins. In yeast, the E3 ligases Doa10 and Hrd1 have similar functions on endoplasmic reticulum proteins.[26] On the molecular level, degradation rate of aggregates vary from protein to protein due to their different internal environments, and thus different accessibility for protease molecules.[27]
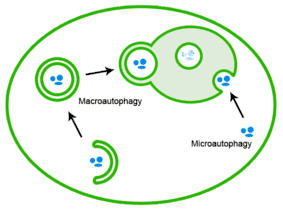
Misfolded proteins can also be eliminated through autophagy, in which the protein aggregates are delivered to the lysosome.[26]
Toxicity
Although it has been thought that the mature protein aggregates themselves are toxic, evidence suggests that it is in fact immature protein aggregates that are most toxic.[28][29] The hydrophobic patches of these aggregates can interact with other components of the cell and damage them. The hypotheses are that the toxicity of protein aggregates is related to mechanisms of the sequestration of cellular components, the generation of reactive oxygen species and the binding to specific receptors in the membrane or through the disruption of membranes.[30] A quantitative assay has been used to determine that higher molecular weight species are responsible for the membrane permeation.[31] It is known that protein aggregates in vitro can destabilize artificial phospholipid bilayers, leading to permeabilization of the membrane.
In biomanufacturing
Protein aggregation is also a common phenomenon in the biopharmaceutical manufacturing process, which may pose risks to patients via generating adverse immune responses.[32]
References
- Aguzzi A, O'Connor T (March 2010). "Protein aggregation diseases: pathogenicity and therapeutic perspectives". Nature Reviews. Drug Discovery. 9 (3): 237–248. doi:10.1038/nrd3050. PMID 20190788. S2CID 5756683.
- Stefani M, Dobson CM (November 2003). "Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution". Journal of Molecular Medicine. 81 (11): 678–699. doi:10.1007/s00109-003-0464-5. PMID 12942175. S2CID 23544974.
- De Felice FG, Vieira MN, Meirelles MN, Morozova-Roche LA, Dobson CM, Ferreira ST (July 2004). "Formation of amyloid aggregates from human lysozyme and its disease-associated variants using hydrostatic pressure". FASEB Journal. 18 (10): 1099–1101. doi:10.1096/fj.03-1072fje. PMID 15155566. S2CID 13647147.
- Tanzi RE, Bertram L (February 2005). "Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective". Cell. 120 (4): 545–555. doi:10.1016/j.cell.2005.02.008. PMID 15734686. S2CID 206559875.
- Brüning A, Jückstock J (2015-01-01). "Misfolded proteins: from little villains to little helpers in the fight against cancer". Frontiers in Oncology. 5: 47. doi:10.3389/fonc.2015.00047. PMC 4338749. PMID 25759792.
- Gething MJ, Sambrook J (January 1992). "Protein folding in the cell". Nature. 355 (6355): 33–45. Bibcode:1992Natur.355...33G. doi:10.1038/355033a0. PMID 1731198. S2CID 4330003.
- Roberts CJ (December 2007). "Non-native protein aggregation kinetics". Biotechnology and Bioengineering. 98 (5): 927–938. doi:10.1002/bit.21627. PMID 17705294. S2CID 21787377.
- Cox DL, Nelson MM (2013). Lehninger Principles of Biochemistry. New York: W.H. Freeman. p. 143. ISBN 978-1-4292-3414-6.
- Palanikumar L, Karpauskaite L, Al-Sayegh M, Chehade I, Alam M, Hassan S, et al. (June 2021). "Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function". Nature Communications. 12 (1): 3962. Bibcode:2021NatCo..12.3962P. doi:10.1038/s41467-021-23985-1. PMC 8233319. PMID 34172723.
- Berke SJ, Paulson HL (June 2003). "Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration". Current Opinion in Genetics & Development. 13 (3): 253–261. doi:10.1016/S0959-437X(03)00053-4. PMID 12787787.
- Grillari J, Grillari-Voglauer R, Jansen-Dürr P (2010). Urbani F, Magariños R, Puertas S, Carretero M, Rodriguez V, Liñares R, et al. (eds.). Post-translational modification of cellular proteins by ubiquitin and ubiquitin-like molecules: role in cellular senescence and aging. Springer Science+Business Media. pp. 172–196. ISBN 978-1-4419-7002-2.
- Weaver RF (2012). Molecular Biology. New York: McGraw-Hill. pp. 122–156, 523–600. ISBN 978-0-07-352532-7.
- Tyedmers J, Mogk A, Bukau B (November 2010). "Cellular strategies for controlling protein aggregation". Nature Reviews. Molecular Cell Biology. 11 (11): 777–788. doi:10.1038/nrm2993. PMID 20944667. S2CID 22449895.
- Stadtman ER, Levine RL (December 2003). "Free radical-mediated oxidation of free amino acids and amino acid residues in proteins". Amino Acids. 25 (3–4): 207–218. doi:10.1007/s00726-003-0011-2. PMID 14661084. S2CID 26844881.
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI (August 2002). "The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans". Proceedings of the National Academy of Sciences of the United States of America. 99 (16): 10417–10422. Bibcode:2002PNAS...9910417M. doi:10.1073/pnas.152161099. PMC 124929. PMID 12122205.
- Coquel AS, Jacob JP, Primet M, Demarez A, Dimiccoli M, Julou T, et al. (April 2013). "Localization of protein aggregation in Escherichia coli is governed by diffusion and nucleoid macromolecular crowding effect". PLOS Computational Biology. 9 (4): e1003038. arXiv:1303.1904. Bibcode:2013PLSCB...9E3038C. doi:10.1371/journal.pcbi.1003038. PMC 3636022. PMID 23633942.
- Scott JJ (January 1987). "The reinnervation of cat muscle spindles by skeletofusimotor axons". Brain Research. 401 (1): 152–154. doi:10.1016/0006-8993(87)91175-9. PMID 2949798. S2CID 42447184.
- Gherardini L, Zajdel A, Pini L, Crimi A (October 2023). "Prediction of misfolded proteins spreading in Alzheimer's disease using machine learning and spreading models". Cerebral Cortex. doi:10.1093/cercor/bhad380. PMID 37833822.
- Bednarska NG, Schymkowitz J, Rousseau F, Van Eldere J (September 2013). "Protein aggregation in bacteria: the thin boundary between functionality and toxicity". Microbiology. 159 (Pt 9): 1795–1806. doi:10.1099/mic.0.069575-0. PMID 23894132.
- Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A (2013-03-08). "Protein aggregation and degradation mechanisms in neurodegenerative diseases". American Journal of Neurodegenerative Disease. 2 (1): 1–14. PMC 3601466. PMID 23516262.
- Wan W, Zeng L, Jin W, Chen X, Shen D, Huang Y, et al. (December 2021). "A Solvatochromic Fluorescent Probe Reveals Polarity Heterogeneity upon Protein Aggregation in Cells". Angewandte Chemie. 60 (49): 25865–25871. doi:10.1002/anie.202107943. PMID 34562048. S2CID 237626399.
- Garcia-Mata R, Gao YS, Sztul E (June 2002). "Hassles with taking out the garbage: aggravating aggresomes". Traffic. 3 (6): 388–396. doi:10.1034/j.1600-0854.2002.30602.x. PMID 12010457. S2CID 305786.
- Gregersen N, Bolund L, Bross P (October 2005). "Protein misfolding, aggregation, and degradation in disease". Molecular Biotechnology. 31 (2): 141–150. doi:10.1385/MB:31:2:141. PMID 16170215. S2CID 36403914.
- Mogk A, Kummer E, Bukau B (2015-01-01). "Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation". Frontiers in Molecular Biosciences. 2: 22. doi:10.3389/fmolb.2015.00022. PMC 4436881. PMID 26042222.
- Liberek K, Lewandowska A, Zietkiewicz S (January 2008). "Chaperones in control of protein disaggregation". The EMBO Journal. 27 (2): 328–335. doi:10.1038/sj.emboj.7601970. PMC 2234349. PMID 18216875.
- Chen B, Retzlaff M, Roos T, Frydman J (August 2011). "Cellular strategies of protein quality control". Cold Spring Harbor Perspectives in Biology. 3 (8): a004374. doi:10.1101/cshperspect.a004374. PMC 3140689. PMID 21746797.
- Wan W, Zeng L, Jin W, Chen X, Shen D, Huang Y, et al. (December 2021). "A Solvatochromic Fluorescent Probe Reveals Polarity Heterogeneity upon Protein Aggregation in Cells". Angewandte Chemie. 60 (49): 25865–25871. doi:10.1002/anie.202107943. PMID 34562048. S2CID 237626399.
- Zhu YJ, Lin H, Lal R (June 2000). "Fresh and nonfibrillar amyloid beta protein(1-40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AbetaP-channel-mediated cellular toxicity". FASEB Journal. 14 (9): 1244–1254. doi:10.1096/fasebj.14.9.1244. PMID 10834946. S2CID 42263619.
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. (September 2001). "The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation". Nature Neuroscience. 4 (9): 887–893. doi:10.1038/nn0901-887. PMID 11528419. S2CID 13516479.
- Soto C (January 2003). "Unfolding the role of protein misfolding in neurodegenerative diseases". Nature Reviews. Neuroscience. 4 (1): 49–60. doi:10.1038/nrn1007. PMID 12511861. S2CID 205499427.
- Flagmeier P, De S, Wirthensohn DC, Lee SF, Vincke C, Muyldermans S, et al. (June 2017). "Ultrasensitive Measurement of Ca2+ Influx into Lipid Vesicles Induced by Protein Aggregates". Angewandte Chemie. 56 (27): 7750–7754. doi:10.1002/anie.201700966. PMC 5615231. PMID 28474754.
- Vázquez-Rey M, Lang DA (July 2011). "Aggregates in monoclonal antibody manufacturing processes". Biotechnology and Bioengineering. 108 (7): 1494–1508. doi:10.1002/bit.23155. PMID 21480193. S2CID 33285577.