Protein–protein interaction
Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.
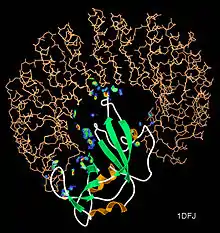
Proteins rarely act alone as their functions tend to be regulated. Many molecular processes within a cell are carried out by molecular machines that are built from numerous protein components organized by their PPIs. These physiological interactions make up the so-called interactomics of the organism, while aberrant PPIs are the basis of multiple aggregation-related diseases, such as Creutzfeldt–Jakob and Alzheimer's diseases.
PPIs have been studied with many methods and from different perspectives: biochemistry, quantum chemistry, molecular dynamics, signal transduction, among others.[1][2][3] All this information enables the creation of large protein interaction networks[4] – similar to metabolic or genetic/epigenetic networks – that empower the current knowledge on biochemical cascades and molecular etiology of disease, as well as the discovery of putative protein targets of therapeutic interest.
Examples
Electron transfer proteins
In many metabolic reactions, a protein that acts as an electron carrier binds to an enzyme that acts as its reductase. After it receives an electron, it dissociates and then binds to the next enzyme that acts as its oxidase (i.e. an acceptor of the electron). These interactions between proteins are dependent on highly specific binding between proteins to ensure efficient electron transfer. Examples: mitochondrial oxidative phosphorylation chain system components cytochrome c-reductase / cytochrome c / cytochrome c oxidase; microsomal and mitochondrial P450 systems.[5]
In the case of the mitochondrial P450 systems, the specific residues involved in the binding of the electron transfer protein adrenodoxin to its reductase were identified as two basic Arg residues on the surface of the reductase and two acidic Asp residues on the adrenodoxin.[6] More recent work on the phylogeny of the reductase has shown that these residues involved in protein–protein interactions have been conserved throughout the evolution of this enzyme.[7]
Signal transduction
The activity of the cell is regulated by extracellular signals. Signal propagation inside and/or along the interior of cells depends on PPIs between the various signaling molecules. The recruitment of signaling pathways through PPIs is called signal transduction and plays a fundamental role in many biological processes and in many diseases including Parkinson's disease and cancer.
Membrane transport
A protein may be carrying another protein (for example, from cytoplasm to nucleus or vice versa in the case of the nuclear pore importins).
Cell metabolism
In many biosynthetic processes enzymes interact with each other to produce small compounds or other macromolecules.
Muscle contraction
Physiology of muscle contraction involves several interactions. Myosin filaments act as molecular motors and by binding to actin enables filament sliding.[8] Furthermore, members of the skeletal muscle lipid droplet-associated proteins family associate with other proteins, as activator of adipose triglyceride lipase and its coactivator comparative gene identification-58, to regulate lipolysis in skeletal muscle
Types
To describe the types of protein–protein interactions (PPIs) it is important to consider that proteins can interact in a "transient" way (to produce some specific effect in a short time, like signal transduction) or to interact with other proteins in a "stable" way to form complexes that become molecular machines within the living systems. A protein complex assembly can result in the formation of homo-oligomeric or hetero-oligomeric complexes. In addition to the conventional complexes, as enzyme-inhibitor and antibody-antigen, interactions can also be established between domain-domain and domain-peptide. Another important distinction to identify protein–protein interactions is the way they have been determined, since there are techniques that measure direct physical interactions between protein pairs, named “binary” methods, while there are other techniques that measure physical interactions among groups of proteins, without pairwise determination of protein partners, named “co-complex” methods.
Homo-oligomers vs. hetero-oligomers
Homo-oligomers are macromolecular complexes constituted by only one type of protein subunit. Protein subunits assembly is guided by the establishment of non-covalent interactions in the quaternary structure of the protein. Disruption of homo-oligomers in order to return to the initial individual monomers often requires denaturation of the complex.[9] Several enzymes, carrier proteins, scaffolding proteins, and transcriptional regulatory factors carry out their functions as homo-oligomers. Distinct protein subunits interact in hetero-oligomers, which are essential to control several cellular functions. The importance of the communication between heterologous proteins is even more evident during cell signaling events and such interactions are only possible due to structural domains within the proteins (as described below).
Stable interactions vs. transient interactions
Stable interactions involve proteins that interact for a long time, taking part of permanent complexes as subunits, in order to carry out functional roles. These are usually the case of homo-oligomers (e.g. cytochrome c), and some hetero-oligomeric proteins, as the subunits of ATPase. On the other hand, a protein may interact briefly and in a reversible manner with other proteins in only certain cellular contexts – cell type, cell cycle stage, external factors, presence of other binding proteins, etc. – as it happens with most of the proteins involved in biochemical cascades. These are called transient interactions. For example, some G protein–coupled receptors only transiently bind to Gi/o proteins when they are activated by extracellular ligands,[10] while some Gq-coupled receptors, such as muscarinic receptor M3, pre-couple with Gq proteins prior to the receptor-ligand binding.[11] Interactions between intrinsically disordered protein regions to globular protein domains (i.e. MoRFs) are transient interactions.[12]
Covalent vs. non-covalent
Covalent interactions are those with the strongest association and are formed by disulphide bonds or electron sharing. While rare, these interactions are determinant in some posttranslational modifications, as ubiquitination and SUMOylation. Non-covalent bonds are usually established during transient interactions by the combination of weaker bonds, such as hydrogen bonds, ionic interactions, Van der Waals forces, or hydrophobic bonds.[13]
Role of water
Water molecules play a significant role in the interactions between proteins.[14][15] The crystal structures of complexes, obtained at high resolution from different but homologous proteins, have shown that some interface water molecules are conserved between homologous complexes. The majority of the interface water molecules make hydrogen bonds with both partners of each complex. Some interface amino acid residues or atomic groups of one protein partner engage in both direct and water mediated interactions with the other protein partner. Doubly indirect interactions, mediated by two water molecules, are more numerous in the homologous complexes of low affinity.[16] Carefully conducted mutagenesis experiments, e.g. changing a tyrosine residue into a phenylalanine, have shown that water mediated interactions can contribute to the energy of interaction.[17] Thus, water molecules may facilitate the interactions and cross-recognitions between proteins.
Structure

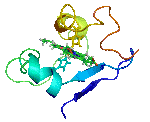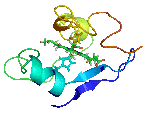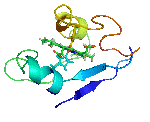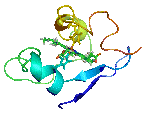
The molecular structures of many protein complexes have been unlocked by the technique of X-ray crystallography.[18][19] The first structure to be solved by this method was that of sperm whale myoglobin by Sir John Cowdery Kendrew.[20] In this technique the angles and intensities of a beam of X-rays diffracted by crystalline atoms are detected in a film, thus producing a three-dimensional picture of the density of electrons within the crystal.[21]
Later, nuclear magnetic resonance also started to be applied with the aim of unravelling the molecular structure of protein complexes. One of the first examples was the structure of calmodulin-binding domains bound to calmodulin.[19][22] This technique is based on the study of magnetic properties of atomic nuclei, thus determining physical and chemical properties of the correspondent atoms or the molecules. Nuclear magnetic resonance is advantageous for characterizing weak PPIs.[23]
Domains
Proteins hold structural domains that allow their interaction with and bind to specific sequences on other proteins:
- Src homology 2 (SH2) domain
- SH2 domains are structurally composed by three-stranded twisted beta sheet sandwiched flanked by two alpha-helices. The existence of a deep binding pocket with high affinity for phosphotyrosine, but not for phosphoserine or phosphothreonine, is essential for the recognition of tyrosine phosphorylated proteins, mainly autophosphorylated growth factor receptors. Growth factor receptor binding proteins and phospholipase Cγ are examples of proteins that have SH2 domains.[24]
- Src homology 3 (SH3) domain
- Structurally, SH3 domains are constituted by a beta barrel formed by two orthogonal beta sheets and three anti-parallel beta strands. These domains recognize proline enriched sequences, as polyproline type II helical structure (PXXP motifs) in cell signaling proteins like protein tyrosine kinases and the growth factor receptor bound protein 2 (Grb2).[24]
- Phosphotyrosine-binding (PTB) domain
- PTB domains interact with sequences that contain a phosphotyrosine group. These domains can be found in the insulin receptor substrate.[24]
- LIM domain
- LIM domains were initially identified in three homeodomain transcription factors (lin11, is11, and mec3). In addition to this homeodomain proteins and other proteins involved in development, LIM domains have also been identified in non-homeodomain proteins with relevant roles in cellular differentiation, association with cytoskeleton and senescence. These domains contain a tandem cysteine-rich Zn2+-finger motif and embrace the consensus sequence CX2CX16-23HX2CX2CX2CX16-21CX2C/H/D. LIM domains bind to PDZ domains, bHLH transcription factors, and other LIM domains.[24]
- Sterile alpha motif (SAM) domain
- SAM domains are composed by five helices forming a compact package with a conserved hydrophobic core. These domains, which can be found in the Eph receptor and the stromal interaction molecule (STIM) for example, bind to non-SAM domain-containing proteins and they also appear to have the ability to bind RNA.[24]
- PDZ domain
- PDZ domains were first identified in three guanylate kinases: PSD-95, DlgA and ZO-1. These domains recognize carboxy-terminal tri-peptide motifs (S/TXV), other PDZ domains or LIM domains and bind them through a short peptide sequence that has a C-terminal hydrophobic residue. Some of the proteins identified as having PDZ domains are scaffolding proteins or seem to be involved in ion receptor assembling and receptor-enzyme complexes formation.[24]
- FERM domain
- FERM domains contain basic residues capable of binding PtdIns(4,5)P2. Talin and focal adhesion kinase (FAK) are two of the proteins that present FERM domains.[24]
- Calponin homology (CH) domain
- Pleckstrin homology domain
- Pleckstrin homology domains bind to phosphoinositides and acid domains in signaling proteins.
- WW domain
- WW domains bind to proline enriched sequences.
- WSxWS motif
- Found in cytokine receptors
Properties of the interface
The study of the molecular structure can give fine details about the interface that enables the interaction between proteins. When characterizing PPI interfaces it is important to take into account the type of complex.[9]
Parameters evaluated include size (measured in absolute dimensions Å2 or in solvent-accessible surface area (SASA)), shape, complementarity between surfaces, residue interface propensities, hydrophobicity, segmentation and secondary structure, and conformational changes on complex formation.[9]
The great majority of PPI interfaces reflects the composition of protein surfaces, rather than the protein cores, in spite of being frequently enriched in hydrophobic residues, particularly in aromatic residues.[25] PPI interfaces are dynamic and frequently planar, although they can be globular and protruding as well.[26] Based on three structures – insulin dimer, trypsin-pancreatic trypsin inhibitor complex, and oxyhaemoglobin – Cyrus Chothia and Joel Janin found that between 1,130 and 1,720 Å2 of surface area was removed from contact with water indicating that hydrophobicity is a major factor of stabilization of PPIs.[27] Later studies refined the buried surface area of the majority of interactions to 1,600±350 Å2. However, much larger interaction interfaces were also observed and were associated with significant changes in conformation of one of the interaction partners.[18] PPIs interfaces exhibit both shape and electrostatic complementarity.[9][11]
Regulation
- Protein concentration, which in turn are affected by expression levels and degradation rates;
- Protein affinity for proteins or other binding ligands;
- Ligands concentrations (substrates, ions, etc.);
- Presence of other proteins, nucleic acids, and ions;
- Electric fields around proteins.
- Occurrence of covalent modifications;
Experimental methods
There are a multitude of methods to detect them.[1][28] Each of the approaches has its own strengths and weaknesses, especially with regard to the sensitivity and specificity of the method. The most conventional and widely used high-throughput methods are yeast two-hybrid screening and affinity purification coupled to mass spectrometry.

Yeast two-hybrid screening
This system was firstly described in 1989 by Fields and Song using Saccharomyces cerevisiae as biological model.[29][30] Yeast two hybrid allows the identification of pairwise PPIs (binary method) in vivo, in which the two proteins are tested for biophysically direct interaction. The Y2H is based on the functional reconstitution of the yeast transcription factor Gal4 and subsequent activation of a selective reporter such as His3. To test two proteins for interaction, two protein expression constructs are made: one protein (X) is fused to the Gal4 DNA-binding domain (DB) and a second protein (Y) is fused to the Gal4 activation domain (AD). In the assay, yeast cells are transformed with these constructs. Transcription of reporter genes does not occur unless bait (DB-X) and prey (AD-Y) interact with each other and form a functional Gal4 transcription factor. Thus, the interaction between proteins can be inferred by the presence of the products resultant of the reporter gene expression.[13][31] In cases in which the reporter gene expresses enzymes that allow the yeast to synthesize essential amino acids or nucleotides, yeast growth under selective media conditions indicates that the two proteins tested are interacting. Recently, software to detect and prioritize protein interactions was published.[32][33]
Despite its usefulness, the yeast two-hybrid system has limitations. It uses yeast as main host system, which can be a problem when studying proteins that contain mammalian-specific post-translational modifications. The number of PPIs identified is usually low because of a high false negative rate;[34] and, understates membrane proteins, for example.[35][36]
In initial studies that utilized Y2H, proper controls for false positives (e.g. when DB-X activates the reporter gene without the presence of AD-Y) were frequently not done, leading to a higher than normal false positive rate. An empirical framework must be implemented to control for these false positives.[37] Limitations in lower coverage of membrane proteins have been overcoming by the emergence of yeast two-hybrid variants, such as the membrane yeast two-hybrid (MYTH)[36] and the split-ubiquitin system,[31] which are not limited to interactions that occur in the nucleus; and, the bacterial two-hybrid system, performed in bacteria;[38]
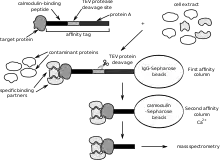
Affinity purification coupled to mass spectrometry
Affinity purification coupled to mass spectrometry mostly detects stable interactions and thus better indicates functional in vivo PPIs.[39][31] This method starts by purification of the tagged protein, which is expressed in the cell usually at in vivo concentrations, and its interacting proteins (affinity purification). One of the most advantageous and widely used methods to purify proteins with very low contaminating background is the tandem affinity purification, developed by Bertrand Seraphin and Matthias Mann and respective colleagues. PPIs can then be quantitatively and qualitatively analysed by mass spectrometry using different methods: chemical incorporation, biological or metabolic incorporation (SILAC), and label-free methods.[9] Furthermore, network theory has been used to study the whole set of identified protein–protein interactions in cells.[4]
Nucleic acid programmable protein array (NAPPA)
This system was first developed by LaBaer and colleagues in 2004 by using in vitro transcription and translation system. They use DNA template encoding the gene of interest fused with GST protein, and it was immobilized in the solid surface. Anti-GST antibody and biotinylated plasmid DNA were bounded in aminopropyltriethoxysilane (APTES)-coated slide. BSA can improve the binding efficiency of DNA. Biotinylated plasmid DNA was bound by avidin. New protein was synthesized by using cell-free expression system i.e. rabbit reticulocyte lysate (RRL), and then the new protein was captured through anti-GST antibody bounded on the slide. To test protein–protein interaction, the targeted protein cDNA and query protein cDNA were immobilized in a same coated slide. By using in vitro transcription and translation system, targeted and query protein was synthesized by the same extract. The targeted protein was bound to array by antibody coated in the slide and query protein was used to probe the array. The query protein was tagged with hemagglutinin (HA) epitope. Thus, the interaction between the two proteins was visualized with the antibody against HA.[40][41]
Intragenic complementation
When multiple copies of a polypeptide encoded by a gene form a complex, this protein structure is referred to as a multimer. When a multimer is formed from polypeptides produced by two different mutant alleles of a particular gene, the mixed multimer may exhibit greater functional activity than the unmixed multimers formed by each of the mutants alone. In such a case, the phenomenon is referred to as intragenic complementation (also called inter-allelic complementation). Intragenic complementation has been demonstrated in many different genes in a variety of organisms including the fungi Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe; the bacterium Salmonella typhimurium; the virus bacteriophage T4,[42] an RNA virus[43] and humans.[44] In such studies, numerous mutations defective in the same gene were often isolated and mapped in a linear order on the basis of recombination frequencies to form a genetic map of the gene. Separately, the mutants were tested in pairwise combinations to measure complementation. An analysis of the results from such studies led to the conclusion that intragenic complementation, in general, arises from the interaction of differently defective polypeptide monomers to form a multimer.[45] Genes that encode multimer-forming polypeptides appear to be common. One interpretation of the data is that polypeptide monomers are often aligned in the multimer in such a way that mutant polypeptides defective at nearby sites in the genetic map tend to form a mixed multimer that functions poorly, whereas mutant polypeptides defective at distant sites tend to form a mixed multimer that functions more effectively. Direct interaction of two nascent proteins emerging from nearby ribosomes appears to be a general mechanism for homo-oligomer (multimer) formation.[46] Hundreds of protein oligomers were identified that assemble in human cells by such an interaction.[46] The most prevalent form of interaction is between the N-terminal regions of the interacting proteins. Dimer formation appears to be able to occur independently of dedicated assembly machines. The intermolecular forces likely responsible for self-recognition and multimer formation were discussed by Jehle.[47]
Other potential methods
Diverse techniques to identify PPIs have been emerging along with technology progression. These include co-immunoprecipitation, protein microarrays, analytical ultracentrifugation, light scattering, fluorescence spectroscopy, luminescence-based mammalian interactome mapping (LUMIER), resonance-energy transfer systems, mammalian protein–protein interaction trap, electro-switchable biosurfaces, protein–fragment complementation assay, as well as real-time label-free measurements by surface plasmon resonance, and calorimetry.[35][36]
Computational methods

Computational prediction of protein–protein interactions
The experimental detection and characterization of PPIs is labor-intensive and time-consuming. However, many PPIs can be also predicted computationally, usually using experimental data as a starting point. However, methods have also been developed that allow the prediction of PPI de novo, that is without prior evidence for these interactions.
Genomic context methods
The Rosetta Stone or Domain Fusion method is based on the hypothesis that interacting proteins are sometimes fused into a single protein in another genome.[48] Therefore, we can predict if two proteins may be interacting by determining if they each have non-overlapping sequence similarity to a region of a single protein sequence in another genome.
The Conserved Neighborhood method is based on the hypothesis that if genes encoding two proteins are neighbors on a chromosome in many genomes, then they are likely functionally related (and possibly physically interacting)[49].
The Phylogenetic Profile method is based on the hypothesis that if two or more proteins are concurrently present or absent across several genomes, then they are likely functionally related.[49] Therefore, potentially interacting proteins can be identified by determining the presence or absence of genes across many genomes and selecting those genes which are always present or absent together.
Text mining methods
Publicly available information from biomedical documents is readily accessible through the internet and is becoming a powerful resource for collecting known protein–protein interactions (PPIs), PPI prediction and protein docking. Text mining is much less costly and time-consuming compared to other high-throughput techniques. Currently, text mining methods generally detect binary relations between interacting proteins from individual sentences using rule/pattern-based information extraction and machine learning approaches.[50] A wide variety of text mining applications for PPI extraction and/or prediction are available for public use, as well as repositories which often store manually validated and/or computationally predicted PPIs. Text mining can be implemented in two stages: information retrieval, where texts containing names of either or both interacting proteins are retrieved and information extraction, where targeted information (interacting proteins, implicated residues, interaction types, etc.) is extracted.
There are also studies using phylogenetic profiling, basing their functionalities on the theory that proteins involved in common pathways co-evolve in a correlated fashion across species. Some more complex text mining methodologies use advanced Natural Language Processing (NLP) techniques and build knowledge networks (for example, considering gene names as nodes and verbs as edges). Other developments involve kernel methods to predict protein interactions.[51]
Machine learning methods
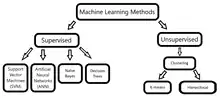
Many computational methods have been suggested and reviewed for predicting protein–protein interactions.[52][53][54] Prediction approaches can be grouped into categories based on predictive evidence: protein sequence, comparative genomics, protein domains, protein tertiary structure, and interaction network topology.[52] The construction of a positive set (known interacting protein pairs) and a negative set (non-interacting protein pairs) is needed for the development of a computational prediction model.[53] Prediction models using machine learning techniques can be broadly classified into two main groups: supervised and unsupervised, based on the labeling of input variables according to the expected outcome.[54]
In 2005, integral membrane proteins of Saccharomyces cerevisiae were analyzed using the mating-based ubiquitin system (mbSUS). The system detects membrane proteins interactions with extracellular signaling proteins[55] Of the 705 integral membrane proteins 1,985 different interactions were traced that involved 536 proteins. To sort and classify interactions a support vector machine was used to define high medium and low confidence interactions. The split-ubiquitin membrane yeast two-hybrid system uses transcriptional reporters to identify yeast transformants that encode pairs of interacting proteins.[56] In 2006, random forest, an example of a supervised technique, was found to be the most-effective machine learning method for protein interaction prediction.[57] Such methods have been applied for discovering protein interactions on human interactome, specifically the interactome of Membrane proteins[58] and the interactome of Schizophrenia-associated proteins.[59]
As of 2020, a model using residue cluster classes (RCCs), constructed from the 3DID and Negatome databases, resulted in 96-99% correctly classified instances of protein–protein interactions.[60] RCCs are a computational vector space that mimics protein fold space and includes all simultaneously contacted residue sets, which can be used to analyze protein structure-function relation and evolution.[61]
Databases
Large scale identification of PPIs generated hundreds of thousands of interactions, which were collected together in specialized biological databases that are continuously updated in order to provide complete interactomes. The first of these databases was the Database of Interacting Proteins (DIP).[62]
Primary databases collect information about published PPIs proven to exist via small-scale or large-scale experimental methods. Examples: DIP, Biomolecular Interaction Network Database (BIND), Biological General Repository for Interaction Datasets (BioGRID), Human Protein Reference Database (HPRD), IntAct Molecular Interaction Database, Molecular Interactions Database (MINT), MIPS Protein Interaction Resource on Yeast (MIPS-MPact), and MIPS Mammalian Protein–Protein Interaction Database (MIPS-MPPI).<
Meta-databases normally result from the integration of primary databases information, but can also collect some original data.
Prediction databases include many PPIs that are predicted using several techniques (main article). Examples: Human Protein–Protein Interaction Prediction Database (PIPs),[63] Interlogous Interaction Database (I2D), Known and Predicted Protein–Protein Interactions (STRING-db), and Unified Human Interactive (UniHI).
The aforementioned computational methods all depend on source databases whose data can be extrapolated to predict novel protein–protein interactions. Coverage differs greatly between databases. In general, primary databases have the fewest total protein interactions recorded as they do not integrate data from multiple other databases, while prediction databases have the most because they include other forms of evidence in addition to experimental. For example, the primary database IntAct has 572,063 interactions,[64] the meta-database APID has 678,000 interactions,[65] and the predictive database STRING has 25,914,693 interactions.[66] However, it is important to note that some of the interactions in the STRING database are only predicted by computational methods such as Genomic Context and not experimentally verified.
Interaction networks
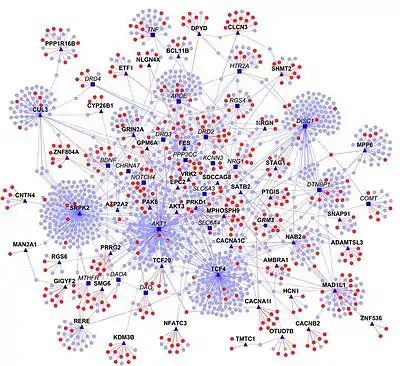
Information found in PPIs databases supports the construction of interaction networks. Although the PPI network of a given query protein can be represented in textbooks, diagrams of whole cell PPIs are frankly complex and difficult to generate.[67]
One example of a manually produced molecular interaction map is the Kurt Kohn's 1999 map of cell cycle control.[68] Drawing on Kohn's map, Schwikowski et al. in 2000 published a paper on PPIs in yeast, linking 1,548 interacting proteins determined by two-hybrid screening. They used a layered graph drawing method to find an initial placement of the nodes and then improved the layout using a force-based algorithm.[69]
Bioinformatic tools have been developed to simplify the difficult task of visualizing molecular interaction networks and complement them with other types of data. For instance, Cytoscape is an open-source software widely used and many plugins are currently available.[70] Pajek software is advantageous for the visualization and analysis of very large networks.[71]
Identification of functional modules in PPI networks is an important challenge in bioinformatics. Functional modules means a set of proteins that are highly connected to each other in PPI network. It is almost similar problem as community detection in social networks. There are some methods such as Jactive[72] modules and MoBaS.[73] Jactive modules integrate PPI network and gene expression data where as MoBaS integrate PPI network and Genome Wide association Studies.
protein–protein relationships are often the result of multiple types of interactions or are deduced from different approaches, including co-localization, direct interaction, suppressive genetic interaction, additive genetic interaction, physical association, and other associations.[74]
Signed interaction networks
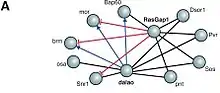
Protein–protein interactions often result in one of the interacting proteins either being 'activated' or 'repressed'. Such effects can be indicated in a PPI network by "signs" (e.g. "activation" or "inhibition"). Although such attributes have been added to networks for a long time,[76] Vinayagam et al. (2014) coined the term Signed network for them. Signed networks are often expressed by labeling the interaction as either positive or negative. A positive interaction is one where the interaction results in one of the proteins being activated. Conversely, a negative interaction indicates that one of the proteins being inactivated.[77]
Protein–protein interaction networks are often constructed as a result of lab experiments such as yeast two-hybrid screens or 'affinity purification and subsequent mass spectrometry techniques.[78] However these methods do not provide the layer of information needed in order to determine what type of interaction is present in order to be able to attribute signs to the network diagrams.
RNA interference screens
RNA interference (RNAi) screens (repression of individual proteins between transcription and translation) are one method that can be utilized in the process of providing signs to the protein–protein interactions. Individual proteins are repressed and the resulting phenotypes are analyzed. A correlating phenotypic relationship (i.e. where the inhibition of either of two proteins results in the same phenotype) indicates a positive, or activating relationship. Phenotypes that do not correlate (i.e. where the inhibition of either of two proteins results in two different phenotypes) indicate a negative or inactivating relationship. If protein A is dependent on protein B for activation then the inhibition of either protein A or B will result in a cell losing the service that is provided by protein A and the phenotypes will be the same for the inhibition of either A or B. If, however, protein A is inactivated by protein B then the phenotypes will differ depending on which protein is inhibited (inhibit protein B and it can no longer inactivate protein A leaving A active however inactivate A and there is nothing for B to activate since A is inactive and the phenotype changes). Multiple RNAi screens need to be performed in order to reliably appoint a sign to a given protein–protein interaction. Vinayagam et al. who devised this technique state that a minimum of nine RNAi screens are required with confidence increasing as one carries out more screens.[77]
As therapeutic targets
Modulation of PPI is challenging and is receiving increasing attention by the scientific community.[79] Several properties of PPI such as allosteric sites and hotspots, have been incorporated into drug-design strategies.[80][81] Nevertheless, very few PPIs are directly targeted by FDA-approved small-molecule PPI inhibitors, emphasizing a huge untapped opportunity for drug discovery.
Recently, Amit Jaiswal and others were able to develop 30 peptides using protein–protein interaction studies to inhibit telomerase recruitment towards telomeres.[82][83]
Examples
- Tirobifan, inhibitor of the glycoprotein IIb/IIIa, used as a cardiovascular drug
- Maraviroc, inhibitor of the CCR5-gp120 interaction, used as anti-HIV drug.[84]
- AMG-176, AZD5991, S64315, inhibitors of myeloid cell leukemia 1 (Mcl-1) protein and its interactions[85]
See also
- Glycan-protein interactions
- 3did
- Allostery
- Biological network
- Biological machines
- DIMA (database)
- Enzyme catalysis
- HitPredict
- Human interactome
- IsoBase
- Multiprotein complex
- Protein domain dynamics
- Protein flexibility
- Protein structure
- Protein–protein interaction prediction
- Protein–protein interaction screening
- Systems biology
References
- Titeca, Kevin; Lemmens, Irma; Tavernier, Jan; Eyckerman, Sven (29 June 2018). "Discovering cellular protein‐protein interactions: Technological strategies and opportunities". Mass Spectrometry Reviews. 38 (1): 79–111. doi:10.1002/mas.21574. ISSN 0277-7037. PMID 29957823.
- Herce HD, Deng W, Helma J, Leonhardt H, Cardoso MC (2013). "Visualization and targeted disruption of protein interactions in living cells". Nature Communications. 4: 2660. Bibcode:2013NatCo...4.2660H. doi:10.1038/ncomms3660. PMC 3826628. PMID 24154492.
- Isa, Nur Firdaus; Bensaude, Olivier; Murphy, Shona (5 February 2022). "Amber Suppression Technology for Mapping Site-specific Viral-host Protein Interactions in Mammalian Cells". Bio-protocol. 12 (3): e4315. doi:10.21769/bioprotoc.4315. PMC 8855090. PMID 35284605.
- Mashaghi, A.; et al. (2004). "Investigation of a protein complex network". European Physical Journal. 41 (1): 113–121. arXiv:cond-mat/0304207. Bibcode:2004EPJB...41..113M. doi:10.1140/epjb/e2004-00301-0. S2CID 9233932.
- Hanukoglu I (1996). "Electron transfer proteins of cytochrome P450 systems". In Bittar EE, Jefcoate CR (eds.). Physiological Functions of Cytochrome P450 in Relation to Structure and Regulation. Advances in Molecular and Cell Biology. Vol. 14. JAI Press, Inc. pp. 29–55. doi:10.1016/S1569-2558(08)60339-2. ISBN 9780762301133.
- Brandt ME, Vickery LE (August 1993). "Charge pair interactions stabilizing ferredoxin-ferredoxin reductase complexes. Identification by complementary site-specific mutations". The Journal of Biological Chemistry. 268 (23): 17126–30. doi:10.1016/S0021-9258(19)85311-5. PMID 8349601.
- Hanukoglu I (2017). "Conservation of the Enzyme-Coenzyme Interfaces in FAD and NADP Binding Adrenodoxin Reductase-A Ubiquitous Enzyme". Journal of Molecular Evolution. 85 (5): 205–218. Bibcode:2017JMolE..85..205H. doi:10.1007/s00239-017-9821-9. PMID 29177972. S2CID 7120148.
- Cooper G (2000). The cell : a molecular approach (2nd ed.). Washington DC: ASM Press. ISBN 978-0-87893-106-4.
- Jones S, Thornton JM (January 1996). "Principles of protein–protein interactions". Proceedings of the National Academy of Sciences of the United States of America. 93 (1): 13–20. Bibcode:1996PNAS...93...13J. doi:10.1073/pnas.93.1.13. PMC 40170. PMID 8552589.
- Qin K, Sethi PR, Lambert NA (August 2008). "Abundance and stability of complexes containing inactive G protein-coupled receptors and G proteins". FASEB Journal. 22 (8): 2920–7. doi:10.1096/fj.08-105775. PMC 2493464. PMID 18434433.
- Qin K, Dong C, Wu G, Lambert NA (August 2011). "Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers". Nature Chemical Biology. 7 (10): 740–7. doi:10.1038/nchembio.642. PMC 3177959. PMID 21873996.
- Malhis N, Gsponer J (June 2015). "Computational identification of MoRFs in protein sequences". Bioinformatics. 31 (11): 1738–44. doi:10.1093/bioinformatics/btv060. PMC 4443681. PMID 25637562.
- Westermarck J, Ivaska J, Corthals GL (July 2013). "Identification of protein interactions involved in cellular signaling". Molecular & Cellular Proteomics. 12 (7): 1752–63. doi:10.1074/mcp.R113.027771. PMC 3708163. PMID 23481661.
- Janin J (December 1999). "Wet and dry interfaces: the role of solvent in protein–protein and protein–DNA recognition". Structure. 7 (12): R277–9. doi:10.1016/s0969-2126(00)88333-1. PMID 10647173.
- Barillari C, Taylor J, Viner R, Essex JW (March 2007). "Classification of water molecules in protein binding sites". Journal of the American Chemical Society. 129 (9): 2577–87. doi:10.1021/ja066980q. PMID 17288418.
- Lisova O, Belkadi L, Bedouelle H (April 2014). "Direct and indirect interactions in the recognition between a cross-neutralizing antibody and the four serotypes of dengue virus". Journal of Molecular Recognition. 27 (4): 205–14. doi:10.1002/jmr.2352. PMID 24591178. S2CID 5416842.
- England P, Brégégère F, Bedouelle H (January 1997). "Energetic and kinetic contributions of contact residues of antibody D1.3 in the interaction with lysozyme". Biochemistry. 36 (1): 164–72. CiteSeerX 10.1.1.613.413. doi:10.1021/bi961419y. PMID 8993330.
- Janin J, Chothia C (September 1990). "The structure of protein–protein recognition sites". The Journal of Biological Chemistry. 265 (27): 16027–30. doi:10.1016/S0021-9258(17)46181-3. PMID 2204619.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002). Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-3218-3.
- Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC (March 1958). "A three-dimensional model of the myoglobin molecule obtained by x-ray analysis". Nature. 181 (4610): 662–6. Bibcode:1958Natur.181..662K. doi:10.1038/181662a0. PMID 13517261. S2CID 4162786.
- Cooper DR, Porebski PJ, Chruszcz M, Minor W (August 2011). "X-ray crystallography: Assessment and validation of protein–small molecule complexes for drug discovery". Expert Opinion on Drug Discovery. 6 (8): 771–782. doi:10.1517/17460441.2011.585154. PMC 3138648. PMID 21779303.
- Wand AJ, Englander SW (August 1996). "Protein complexes studied by NMR spectroscopy". Current Opinion in Biotechnology. 7 (4): 403–8. doi:10.1016/s0958-1669(96)80115-7. PMC 3442359. PMID 8768898.
- Vinogradova O, Qin J (2012). "NMR as a unique tool in assessment and complex determination of weak protein–protein interactions". In Zhu G (ed.). NMR of Proteins and Small Biomolecules. Topics in Current Chemistry. Vol. 326. Springer Berlin. pp. 35–45. doi:10.1007/128_2011_216. ISBN 978-3-642-28916-3. PMC 3676910. PMID 21809187.
- Berridge, M.J. (2012). "Cell Signalling Biology: Module 6 – Spatial and Temporal Aspects of Signalling". Biochemical Journal. 6: csb0001006. doi:10.1042/csb0001006.
- Yan C, Wu F, Jernigan RL, Dobbs D, Honavar V (January 2008). "Characterization of protein–protein interfaces". The Protein Journal. 27 (1): 59–70. doi:10.1007/s10930-007-9108-x. PMC 2566606. PMID 17851740.
- Jones S, Thornton JM (September 1997). "Analysis of protein–protein interaction sites using surface patches". Journal of Molecular Biology. 272 (1): 121–32. doi:10.1006/jmbi.1997.1234. PMID 9299342.
- Chothia C, Janin J (August 1975). "Principles of protein–protein recognition". Nature. 256 (5520): 705–8. Bibcode:1975Natur.256..705C. doi:10.1038/256705a0. PMID 1153006. S2CID 4292325.
- Phizicky EM, Fields S (March 1995). "protein–protein interactions: methods for detection and analysis". Microbiological Reviews. 59 (1): 94–123. doi:10.1128/MMBR.59.1.94-123.1995. PMC 239356. PMID 7708014.
- Pagel, P.; Kovac, S.; Oesterheld, M.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Mark, P.; Stumpflen, V.; Mewes, H.-W.; Ruepp, A.; Frishman, D. (2005). "The MIPS mammalian protein–protein interaction database". Bioinformatics. 21 (6): 832–834. doi:10.1093/bioinformatics/bti115. PMID 15531608. Retrieved 2 January 2021.
- Terentiev AA, Moldogazieva NT, Shaitan KV (December 2009). "Dynamic proteomics in modeling of the living cell. protein–protein interactions". Biochemistry. Biokhimiia. 74 (13): 1586–607. doi:10.1134/s0006297909130112. PMID 20210711. S2CID 19815231.
- Wodak SJ, Vlasblom J, Turinsky AL, Pu S (December 2013). "protein–protein interaction networks: the puzzling riches". Current Opinion in Structural Biology. 23 (6): 941–53. doi:10.1016/j.sbi.2013.08.002. PMID 24007795.
- Banerjee S, Velásquez-Zapata V, Fuerst G, Elmore JM, Wise RP (December 2020). "NGPINT: a next-generation protein–protein interaction software". Briefings in Bioinformatics. 22 (4). doi:10.1093/bib/bbaa351. PMID 33367498.
- Velásquez-Zapata V, Elmore JM, Banerjee S, Dorman K, Wise RP (April 2021). "Next-generation yeast-two-hybrid analysis with Y2H-SCORES identifies novel interactors of the MLA immune receptor". PLOS Computational Biology. 23 (4): e1008890. Bibcode:2021PLSCB..17E8890V. doi:10.1371/journal.pcbi.1008890. PMC 8046355. PMID 33798202.
- Rajagopala SV, Sikorski P, Caufield JH, Tovchigrechko A, Uetz P (December 2012). "Studying protein complexes by the yeast two-hybrid system". Methods. 58 (4): 392–9. doi:10.1016/j.ymeth.2012.07.015. PMC 3517932. PMID 22841565.
- Stelzl U, Wanker EE (December 2006). "The value of high quality protein–protein interaction networks for systems biology". Current Opinion in Chemical Biology. 10 (6): 551–8. doi:10.1016/j.cbpa.2006.10.005. PMID 17055769.
- Petschnigg J, Snider J, Stagljar I (February 2011). "Interactive proteomics research technologies: recent applications and advances". Current Opinion in Biotechnology. 22 (1): 50–8. doi:10.1016/j.copbio.2010.09.001. PMID 20884196.
- Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabási AL, Vidal M (2009). "An empirical framework for binary interactome mapping". Nat Methods. 6 (1): 83–90. doi:10.1038/nmeth.1280. PMC 2872561. PMID 19060904.
- Battesti A, Bouveret E (December 2012). "The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli". Methods. 58 (4): 325–34. doi:10.1016/j.ymeth.2012.07.018. PMID 22841567.
- Brettner LM, Masel J (September 2012). "Protein stickiness, rather than number of functional protein–protein interactions, predicts expression noise and plasticity in yeast". BMC Systems Biology. 6: 128. doi:10.1186/1752-0509-6-128. PMC 3527306. PMID 23017156.
- Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, Lau AY, Walter JC, LaBaer J (July 2004). "Self-assembling protein microarrays". Science. 305 (5680): 86–90. Bibcode:2004Sci...305...86R. doi:10.1126/science.1097639. PMID 15232106. S2CID 20936301.
- Ramachandran N, Raphael JV, Hainsworth E, Demirkan G, Fuentes MG, Rolfs A, Hu Y, LaBaer J (June 2008). "Next-generation high-density self-assembling functional protein arrays". Nature Methods. 5 (6): 535–8. doi:10.1038/nmeth.1210. PMC 3070491. PMID 18469824.
- Bernstein, H; Edgar, RS; Denhardt, GH (June 1965). "Intragenic Complementation among Temperature Sensitive Mutants of Bacteriophage T4D". Genetics. 51 (6): 987–1002. doi:10.1093/genetics/51.6.987. PMC 1210828. PMID 14337770.
- Smallwood S, Cevik B, Moyer SA. Intragenic complementation and oligomerization of the L subunit of the sendai virus RNA polymerase. Virology. 2002;304(2):235-245. doi:10.1006/viro.2002.1720
- Rodríguez-Pombo P, Pérez-Cerdá C, Pérez B, Desviat LR, Sánchez-Pulido L, Ugarte M. Towards a model to explain the intragenic complementation in the heteromultimeric protein propionyl-CoA carboxylase. Biochim Biophys Acta. 2005;1740(3):489-498. doi:10.1016/j.bbadis.2004.10.009
- Crick FH, Orgel LE. The theory of inter-allelic complementation. J Mol Biol. 1964 Jan;8:161-5. doi:10.1016/s0022-2836(64)80156-x. PMID 14149958
- Bertolini M, Fenzl K, Kats I, Wruck F, Tippmann F, Schmitt J, Auburger JJ, Tans S, Bukau B, Kramer G. Interactions between nascent proteins translated by adjacent ribosomes drive homomer assembly. Science. 2021 Jan 1;371(6524):57-64. doi:10.1126/science.abc7151. PMID 33384371
- Jehle H. Intermolecular forces and biological specificity. Proc Natl Acad Sci U S A 1963;50(3):516-524. doi:10.1073/pnas.50.3.516
- Eisenberg, David; Yeates, Todd O.; Rice, Danny W.; Ng, Ho-Leung; Pellegrini, Matteo; Marcotte, Edward M. (30 July 1999). "Detecting Protein Function and protein–Protein Interactions from Genome Sequences". Science. 285 (5428): 751–753. CiteSeerX 10.1.1.535.9650. doi:10.1126/science.285.5428.751. ISSN 1095-9203. PMID 10427000.
- Raman, Karthik (15 February 2010). "Construction and analysis of protein–protein interaction networks". Automated Experimentation. 2 (1): 2. doi:10.1186/1759-4499-2-2. ISSN 1759-4499. PMC 2834675. PMID 20334628.
- Badal VD, Kundrotas PJ, Vakser IA (December 2015). "Text Mining for Protein Docking". PLOS Computational Biology. 11 (12): e1004630. Bibcode:2015PLSCB..11E4630B. doi:10.1371/journal.pcbi.1004630. PMC 4674139. PMID 26650466.
- Papanikolaou N, Pavlopoulos GA, Theodosiou T, Iliopoulos I (March 2015). "protein–protein interaction predictions using text mining methods". Methods. Text mining of biomedical literature. 74: 47–53. doi:10.1016/j.ymeth.2014.10.026. PMID 25448298.
- Kotlyar, Max; Rossos, Andrea E.M.; Jurisica, Igor (December 2017). "Prediction of Protein‐Protein Interactions". Current Protocols in Bioinformatics. 60 (1): 8.2.1–8.2.14. doi:10.1002/cpbi.38. PMID 29220074. S2CID 19509320.
- Ding, Ziyun; Kihara, Daisuke (August 2018). "Computational Methods for Predicting protein–Protein Interactions Using Various Protein Features". Current Protocols in Protein Science. 93 (1): e62. doi:10.1002/cpps.62. PMC 6097941. PMID 29927082.
- Sarkar, Debasree; Saha, Sudipto (September 2019). "Machine-learning techniques for the prediction of protein–protein interactions". Journal of Biosciences. 44 (4): 104. doi:10.1007/s12038-019-9909-z. PMID 31502581. S2CID 199668359.
- Miller, John P.; Lo, Russell S.; Ben-Hur, Asa; Desmarais, Cynthia; Stagljar, Igor; Noble, William Stafford; Fields, Stanley (2005). "Large-scale identification of yeast integral membrane protein interactions". Proceedings of the National Academy of Sciences of the United States of America. 102 (34): 12123–12128. Bibcode:2005PNAS..10212123M. doi:10.1073/pnas.0505482102. PMC 1189342. PMID 16093310.
- Sylvie Lalonde, Antoinette Sero, Réjane Pratelli, Guillaume Pilot, Jin Chen, Maria I. Sardi, Saman A. Parsa, Do-Young Kim, Biswa R. Acharya, Erica V. Stein, Heng-Chen Hu, Florent Villiers, Kouji Takeda, Yingzhen Yang, Yong S. Han, Rainer Schwacke, William Chiang, Naohiro Kato, Dominique Loqué, Sarah M. Assmann, June M. Kwak, Julian I. Schroeder, Seung Y. Rhee and Wolf B. Frommer (2010). "A membrane protein/signaling protein interaction network for Arabidopsis version AMPv2". Frontiers in Physiology. 1: 24. doi:10.3389/fphys.2010.00024. PMC 3059934. PMID 21423366.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Qi Y, Bar-Joseph Z, Klein-Seetharaman J (May 2006). "Evaluation of different biological data and computational classification methods for use in protein interaction prediction". Proteins. 63 (3): 490–500. doi:10.1002/prot.20865. PMC 3250929. PMID 16450363.
- Qi Y, Dhiman HK, Bhola N, Budyak I, Kar S, Man D, Dutta A, Tirupula K, Carr BI, Grandis J, Bar-Joseph Z, Klein-Seetharaman J (December 2009). "Systematic prediction of human membrane receptor interactions". Proteomics. 9 (23): 5243–55. doi:10.1002/pmic.200900259. PMC 3076061. PMID 19798668.
- Ganapathiraju MK, Thahir M, Handen A, Sarkar SN, Sweet RA, Nimgaonkar VL, Loscher CE, Bauer EM, Chaparala S (April 2016). "Schizophrenia interactome with 504 novel protein–protein interactions". npj Schizophrenia. 2: 16012. doi:10.1038/npjschz.2016.12. PMC 4898894. PMID 27336055.
- Poot Velez, Albros Hermes; Fontove, Fernando; Del Rio, Gabriel (6 July 2020). "Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes". International Journal of Molecular Sciences. 21 (13): 4787. doi:10.3390/ijms21134787. PMC 7370293. PMID 32640745.
- Corral-Corral, Ricardo; Chavez, Edgar; Del Rio, Gabriel (December 2015). "Machine Learnable Fold Space Representation based on Residue Cluster Classes". Computational Biology and Chemistry. 59: 1–7. doi:10.1016/j.compbiolchem.2015.07.010. PMID 26366526.
- Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D (January 2000). "DIP: the database of interacting proteins". Nucleic Acids Research. 28 (1): 289–91. doi:10.1093/nar/28.1.289. PMC 102387. PMID 10592249.
- McDowall MD, Scott MS, Barton GJ (January 2009). "PIPs: human protein–protein interaction prediction database". Nucleic Acids Research. 37 (Database issue): D651–6. doi:10.1093/nar/gkn870. PMC 2686497. PMID 18988626.
- IntAct. "Proteins, Interactions, Binary interactions and n-ary interactions". www.ebi.ac.uk. Retrieved 19 November 2018.
- "Agile Protein Interactomes DataServer: About APID".
- "STRING: functional protein association networks". string-db.org. Retrieved 19 November 2018.
- Sprinzak, Einat; Sattath, Shmuel; Margalit, Hanah (2003). "How Reliable are Experimental Protein–Protein Interaction Data?". Journal of Molecular Biology. 327 (5): 919–923. doi:10.1016/S0022-2836(03)00239-0. PMID 12662919. Retrieved 2 January 2021.
- Schwikowski B, Uetz P, Fields S (December 2000). "A network of protein–protein interactions in yeast". Nature Biotechnology. 18 (12): 1257–61. doi:10.1038/82360. PMID 11101803. S2CID 3009359.
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B (October 1999). "A generic protein purification method for protein complex characterization and proteome exploration". Nature Biotechnology. 17 (10): 1030–2. doi:10.1038/13732. PMID 10504710. S2CID 663553.
- Kohl M, Wiese S, Warscheid B (2011). "Cytoscape: Software for Visualization and Analysis of Biological Networks". Data Mining in Proteomics. Methods in Molecular Biology. Vol. 696. pp. 291–303. doi:10.1007/978-1-60761-987-1_18. ISBN 978-1-60761-986-4. PMID 21063955.
- Raman K (February 2010). "Construction and analysis of protein–protein interaction networks". Automated Experimentation. 2 (1): 2. doi:10.1186/1759-4499-2-2. PMC 2834675. PMID 20334628.
- Ideker T, Ozier O, Schwikowski B, Siegel AF (1 January 2002). "Discovering regulatory and signalling circuits in molecular interaction networks". Bioinformatics. 18 (Suppl 1): S233–40. doi:10.1093/bioinformatics/18.suppl_1.s233. PMID 12169552.
- Ayati M, Erten S, Chance MR, Koyutürk M (30 June 2015). "MOBAS: identification of disease-associated protein subnetworks using modularity-based scoring". EURASIP Journal on Bioinformatics and Systems Biology. 2015 (1): 7. doi:10.1186/s13637-015-0025-6. ISSN 1687-4153. PMC 5270451. PMID 28194175.
- De Domenico M, Nicosia V, Arenas A, Latora V (April 2015). "Structural reducibility of multilayer networks". Nature Communications. 6: 6864. arXiv:1405.0425. Bibcode:2015NatCo...6.6864D. doi:10.1038/ncomms7864. PMID 25904309. S2CID 16776349.
- Fischer B, Sandmann T, Horn T, Billmann M, Chaudhary V, Huber W, Boutros M (March 2015). "A map of directional genetic interactions in a metazoan cell". eLife. 4. doi:10.7554/eLife.05464. PMC 4384530. PMID 25748138.
- Ideker T., Tan K. & Uetz P. (2005) Visualization and integration of protein–protein interactions. In: Golemis, E. (ed.) protein–Protein Interactions – A Molecular Cloning Manual, 2nd ed. Cold Spring Harbor Laboratory Press.
- Vinayagam A, Zirin J, Roesel C, Hu Y, Yilmazel B, Samsonova AA, Neumüller RA, Mohr SE, Perrimon N (January 2014). "Integrating protein–protein interaction networks with phenotypes reveals signs of interactions". Nature Methods. 11 (1): 94–9. doi:10.1038/nmeth.2733. PMC 3877743. PMID 24240319.
- Chen GI, Gingras AC (July 2007). "Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases". Methods. 42 (3): 298–305. doi:10.1016/j.ymeth.2007.02.018. PMID 17532517.
- Laraia L, McKenzie G, Spring DR, Venkitaraman AR, Huggins DJ (June 2015). "Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting protein–Protein Interactions". Chemistry & Biology. 22 (6): 689–703. doi:10.1016/j.chembiol.2015.04.019. PMC 4518475. PMID 26091166.
- Arkin MR, Wells JA (April 2004). "Small-molecule inhibitors of protein–protein interactions: progressing towards the dream". Nature Reviews. Drug Discovery. 3 (4): 301–17. doi:10.1038/nrd1343. PMC 4179228. PMID 15060526. S2CID 13879559.
- Chen J, Sawyer N, Regan L (April 2013). "protein–protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area". Protein Science. 22 (4): 510–5. doi:10.1002/pro.2230. PMC 3610057. PMID 23389845.
- Jaiswal A, Lakshmi PT (9 September 2014). "Molecular inhibition of telomerase recruitment using designer peptides: an in silico approach". Journal of Biomolecular Structure & Dynamics. 33 (7): 1442–59. doi:10.1080/07391102.2014.953207. PMID 25204447. S2CID 27293727.
- Jaiswal A (2014). "AtTRB1–3 Mediates Structural Changes in AtPOT1b to Hold ssDNA". ISRN Structural Biology. 2014: 1–16. doi:10.1155/2014/827201.
- Ivanov AA, Khuri FR, Fu H (July 2013). "Targeting protein–protein interactions as an anticancer strategy". Trends in Pharmacological Sciences. 34 (7): 393–400. doi:10.1016/j.tips.2013.04.007. PMC 3773978. PMID 23725674.
- Hargreaves, David; Carbajo, Rodrigo J.; Bodnarchuk, Michael S.; Embrey, Kevin; Rawlins, Philip B.; Packer, Martin; Degorce, Sébastien L.; Hird, Alexander W.; Johannes, Jeffrey W.; Chiarparin, Elisabetta; Schade, Markus (23 May 2023). "Design of rigid protein–protein interaction inhibitors enables targeting of undruggable Mcl-1". Proceedings of the National Academy of Sciences. 120 (21): e2221967120. doi:10.1073/pnas.2221967120. ISSN 0027-8424. PMC 10214187. PMID 37186857.
Further reading
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M (January 2006). "BioGRID: a general repository for interaction datasets". Nucleic Acids Research. 34 (Database issue): D535–9. doi:10.1093/nar/gkj109. PMC 1347471. PMID 16381927.
- Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN, Deshpande N, Suresh S, Rashmi BP, Shanker K, Padma N, Niranjan V, Harsha HC, Talreja N, Vrushabendra BM, Ramya MA, Yatish AJ, Joy M, Shivashankar HN, Kavitha MP, Menezes M, Choudhury DR, Ghosh N, Saravana R, Chandran S, Mohan S, Jonnalagadda CK, Prasad CK, Kumar-Sinha C, Deshpande KS, Pandey A (January 2004). "Human protein reference database as a discovery resource for proteomics". Nucleic Acids Research. 32 (Database issue): D497–501. doi:10.1093/nar/gkh070. PMC 308804. PMID 14681466.
- Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, Vingron M, Roechert B, Roepstorff P, Valencia A, Margalit H, Armstrong J, Bairoch A, Cesareni G, Sherman D, Apweiler R (January 2004). "IntAct: an open source molecular interaction database". Nucleic Acids Research. 32 (Database issue): D452–5. doi:10.1093/nar/gkh052. PMC 308786. PMID 14681455.
- Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G (January 2007). "MINT: the Molecular INTeraction database". Nucleic Acids Research. 35 (Database issue): D572–4. doi:10.1093/nar/gkl950. PMC 1751541. PMID 17135203.
- Güldener U, Münsterkötter M, Oesterheld M, Pagel P, Ruepp A, Mewes HW, Stümpflen V (January 2006). "MPact: the MIPS protein interaction resource on yeast". Nucleic Acids Research. 34 (Database issue): D436–41. doi:10.1093/nar/gkj003. PMC 1347366. PMID 16381906.
- Pagel P, Kovac S, Oesterheld M, Brauner B, Dunger-Kaltenbach I, Frishman G, Montrone C, Mark P, Stümpflen V, Mewes HW, Ruepp A, Frishman D (March 2005). "The MIPS mammalian protein–protein interaction database". Bioinformatics. 21 (6): 832–4. doi:10.1093/bioinformatics/bti115. PMID 15531608.
External links
- Protein–Protein Interaction Databases
- Library of Modulators of Protein–Protein Interactions (PPI)
- Proteins and Enzymes at Curlie
- Casado-Vela J, Matthiesen R, Sellés S, Naranjo JR (May 2013). "Protein–Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration". Proteomes. 1 (1): 3–24. doi:10.3390/proteomes1010003. PMC 5314489. PMID 28250396.
- Robin V, Bodein A, Scott-Boyer MP, Leclercq M, Périn O, Droit A. Overview of methods for characterization and visualization of a protein–protein interaction network in a multi-omics integration context. Front Mol Biosci. 2022 Sep 8;9:962799. doi: 10.3389/fmolb.2022.962799. PMID 36158572; PMCID: PMC9494275.
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology | |
| Research tools | |
| Organizations | |
| |