Protein dynamics
Proteins are generally thought to adopt unique structures determined by their amino acid sequences. However, proteins are not strictly static objects, but rather populate ensembles of (sometimes similar) conformations. Transitions between these states occur on a variety of length scales (tenths of Å to nm) and time scales (ns to s), and have been linked to functionally relevant phenomena such as allosteric signaling[1] and enzyme catalysis.[2]
The study of protein dynamics is most directly concerned with the transitions between these states, but can also involve the nature and equilibrium populations of the states themselves. These two perspectives—kinetics and thermodynamics, respectively—can be conceptually synthesized in an "energy landscape" paradigm:[3] highly populated states and the kinetics of transitions between them can be described by the depths of energy wells and the heights of energy barriers, respectively.

Local flexibility: atoms and residues
Portions of protein structures often deviate from the equilibrium state. Some such excursions are harmonic, such as stochastic fluctuations of chemical bonds and bond angles. Others are anharmonic, such as sidechains that jump between separate discrete energy minima, or rotamers.[4]
Evidence for local flexibility is often obtained from NMR spectroscopy. Flexible and potentially disordered regions of a protein can be detected using the random coil index. Flexibility in folded proteins can be identified by analyzing the spin relaxation of individual atoms in the protein. Flexibility can also be observed in very high-resolution electron density maps produced by X-ray crystallography,[5] particularly when diffraction data is collected at room temperature instead of the traditional cryogenic temperature (typically near 100 K).[6] Information on the frequency distribution and dynamics of local protein flexibility can be obtained using Raman and optical Kerr-effect spectroscopy[7] as well as anisotropic microspectroscopy[8] in the terahertz frequency domain.
Regional flexibility: intra-domain multi-residue coupling
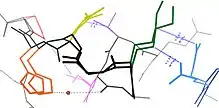
Many residues are in close spatial proximity in protein structures. This is true for most residues that are contiguous in the primary sequence, but also for many that are distal in sequence yet are brought into contact in the final folded structure. Because of this proximity, these residue's energy landscapes become coupled based on various biophysical phenomena such as hydrogen bonds, ionic bonds, and van der Waals interactions (see figure).
Transitions between states for such sets of residues therefore become correlated.[9]
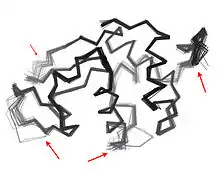
This is perhaps most obvious for surface-exposed loops, which often shift collectively to adopt different conformations in different crystal structures (see figure). However, coupled conformational heterogeneity is also sometimes evident in secondary structure.[10] For example, consecutive residues and residues offset by 4 in the primary sequence often interact in α helices. Also, residues offset by 2 in the primary sequence point their sidechains toward the same face of β sheets and are close enough to interact sterically, as are residues on adjacent strands of the same β sheet. Some of these conformational changes are induced by post-translational modifications in protein structure, such as phosphorylation and methylation.[10][11]
When these coupled residues form pathways linking functionally important parts of a protein, they may participate in allosteric signaling. For example, when a molecule of oxygen binds to one subunit of the hemoglobin tetramer, that information is allosterically propagated to the other three subunits, thereby enhancing their affinity for oxygen. In this case, the coupled flexibility in hemoglobin allows for cooperative oxygen binding, which is physiologically useful because it allows rapid oxygen loading in lung tissue and rapid oxygen unloading in oxygen-deprived tissues (e.g. muscle).
Global flexibility: multiple domains
The presence of multiple domains in proteins gives rise to a great deal of flexibility and mobility, leading to protein domain dynamics.[1] Domain motions can be inferred by comparing different structures of a protein (as in Database of Molecular Motions), or they can be directly observed using spectra[12][13] measured by neutron spin echo spectroscopy. They can also be suggested by sampling in extensive molecular dynamics trajectories[14] and principal component analysis.[15] Domain motions are important for:
- ABC transporters [16]
- catalysis[17]
- cellular locomotion and motor proteins[18]
- formation of protein complexes[19]
- ion channels[20]
- mechanoreceptors and mechanotransduction[21]
- regulatory activity [22]
- transport of metabolites across cell membranes
One of the largest observed domain motions is the 'swivelling' mechanism in pyruvate phosphate dikinase. The phosphoinositide domain swivels between two states in order to bring a phosphate group from the active site of the nucleotide binding domain to that of the phosphoenolpyruvate/pyruvate domain.[23] The phosphate group is moved over a distance of 45 Å involving a domain motion of about 100 degrees around a single residue. In enzymes, the closure of one domain onto another captures a substrate by an induced fit, allowing the reaction to take place in a controlled way. A detailed analysis by Gerstein led to the classification of two basic types of domain motion; hinge and shear.[20] Only a relatively small portion of the chain, namely the inter-domain linker and side chains undergo significant conformational changes upon domain rearrangement.[24]
Hinge motions
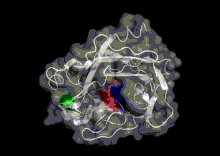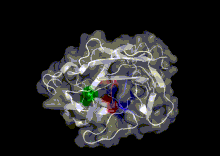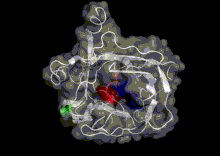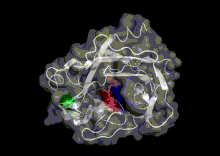
A study by Hayward[25] found that the termini of α-helices and β-sheets form hinges in a large number of cases. Many hinges were found to involve two secondary structure elements acting like hinges of a door, allowing an opening and closing motion to occur. This can arise when two neighbouring strands within a β-sheet situated in one domain, diverge apart as they join the other domain. The two resulting termini then form the bending regions between the two domains. α-helices that preserve their hydrogen bonding network when bent are found to behave as mechanical hinges, storing `elastic energy' that drives the closure of domains for rapid capture of a substrate.[25] Khade et. al. worked on prediction of the hinges[26] in any conformation and further built an Elastic Network Model called hdANM[27] that can model those motions.
Helical to extended conformation
The interconversion of helical and extended conformations at the site of a domain boundary is not uncommon. In calmodulin, torsion angles change for five residues in the middle of a domain linking α-helix. The helix is split into two, almost perpendicular, smaller helices separated by four residues of an extended strand.[28][29]
Shear motions
Shear motions involve a small sliding movement of domain interfaces, controlled by the amino acid side chains within the interface. Proteins displaying shear motions often have a layered architecture: stacking of secondary structures. The interdomain linker has merely the role of keeping the domains in close proximity.
Domain motion and functional dynamics in enzymes
The analysis of the internal dynamics of structurally different, but functionally similar enzymes has highlighted a common relationship between the positioning of the active site and the two principal protein sub-domains. In fact, for several members of the hydrolase superfamily, the catalytic site is located close to the interface separating the two principal quasi-rigid domains.[14] Such positioning appears instrumental for maintaining the precise geometry of the active site, while allowing for an appreciable functionally oriented modulation of the flanking regions resulting from the relative motion of the two sub-domains.
Implications for macromolecular evolution
Evidence suggests that protein dynamics are important for function, e.g. enzyme catalysis in DHFR, yet they are also posited to facilitate the acquisition of new functions by molecular evolution.[30] This argument suggests that proteins have evolved to have stable, mostly unique folded structures, but the unavoidable residual flexibility leads to some degree of functional promiscuity, which can be amplified/harnessed/diverted by subsequent mutations.
However, there is growing awareness that intrinsically unstructured proteins are quite prevalent in eukaryotic genomes,[31] casting further doubt on the simplest interpretation of Anfinsen's dogma: "sequence determines structure (singular)". In effect, the new paradigm is characterized by the addition of two caveats: "sequence and cellular environment determine structural ensemble".
References
- Bu Z, Callaway DJ (2011). "Proteins move! Protein dynamics and long-range allostery in cell signaling". Protein Structure and Diseases. pp. 163–221. doi:10.1016/B978-0-12-381262-9.00005-7. ISBN 9780123812629. PMID 21570668.
{{cite book}}:|journal=ignored (help) - Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T (Dec 2009). "Hidden alternative structures of proline isomerase essential for catalysis". Nature. 462 (7273): 669–673. Bibcode:2009Natur.462..669F. doi:10.1038/nature08615. PMC 2805857. PMID 19956261.
- Frauenfelder H, Sligar SG, Wolynes PG (Dec 1991). "The energy landscapes and motions of proteins". Science. 254 (5038): 1598–1603. Bibcode:1991Sci...254.1598F. doi:10.1126/science.1749933. PMID 1749933.
- Dunbrack, Roland L (August 2002). "Rotamer Libraries in the 21st Century". Current Opinion in Structural Biology. 12 (4): 431–440. doi:10.1016/s0959-440x(02)00344-5. PMID 12163064.
- Davis IW, Arendall WB, Richardson DC, Richardson JS (Feb 2006). "The backrub motion: how protein backbone shrugs when a sidechain dances". Structure. 14 (2): 265–274. doi:10.1016/j.str.2005.10.007. PMID 16472746.
- Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T (Sep 2011). "Accessing protein conformational ensembles using room-temperature X-ray crystallography". Proceedings of the National Academy of Sciences of the United States of America. 108 (39): 16247–16252. Bibcode:2011PNAS..10816247F. doi:10.1073/pnas.1111325108. PMC 3182744. PMID 21918110.
- Turton DA, Senn HM, Harwood T, Lapthorn AJ, Ellis EM, Wynne K (June 2014). "Terahertz underdamped vibrational motion governs protein-ligand binding in solution". Nature Communications. 5: 3999. Bibcode:2014NatCo...5.3999T. doi:10.1038/ncomms4999. PMID 24893252.
- Acbas, G.; Niessen, K. A.; Snell, E. H.; Markelz, A. G. (2014). "Protein Optical measurements of long-range protein vibrations". Nature Communications. 5: 3076. doi:10.1038/ncomms4076. PMID 24430203.
- Bu Z, Cook J, Callaway DJ (Sep 2001). "Dynamic regimes and correlated structural dynamics in native and denatured alpha-lactalbumin". Journal of Molecular Biology. 312 (4): 865–873. doi:10.1006/jmbi.2001.5006. PMID 11575938.
- Costa CH, Oliveira AR, Dos Santos AM, da Costa KS, Lima AH, Alves CN, Lameira J (October 2019). "Computational study of conformational changes in human 3-hydroxy-3-methylglutaryl coenzyme reductase induced by substrate binding". Journal of Biomolecular Structure & Dynamics. 37 (16): 4374–4383. doi:10.1080/07391102.2018.1549508. PMID 30470158. S2CID 53717806.
- Groban ES, Narayanan A, Jacobson MP (April 2006). Shakhnovich E (ed.). "Conformational changes in protein loops and helices induced by post-translational phosphorylation". PLOS Computational Biology. 2 (4): e32. Bibcode:2006PLSCB...2...32G. doi:10.1371/journal.pcbi.0020032. PMC 1440919. PMID 16628247.
- Farago B, Li J, Cornilescu G, Callaway DJ, Bu Z (Nov 2010). "Activation of nanoscale allosteric protein domain motion revealed by neutron spin echo spectroscopy". Biophysical Journal. 99 (10): 3473–3482. Bibcode:2010BpJ....99.3473F. doi:10.1016/j.bpj.2010.09.058. PMC 2980739. PMID 21081097.
- Bu Z, Biehl R, Monkenbusch M, Richter D, Callaway DJ (Dec 2005). "Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy" (PDF). Proceedings of the National Academy of Sciences of the United States of America. 102 (49): 17646–17651. Bibcode:2005PNAS..10217646B. doi:10.1073/pnas.0503388102. PMC 1345721. PMID 16306270.
- Potestio R, Pontiggia F, Micheletti C (Jun 2009). "Coarse-grained description of protein internal dynamics: an optimal strategy for decomposing proteins in rigid subunits". Biophysical Journal. 96 (12): 4993–5002. Bibcode:2009BpJ....96.4993P. doi:10.1016/j.bpj.2009.03.051. PMC 2712024. PMID 19527659.
- Baron R, Vellore NA (Jul 2012). "LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition". Proceedings of the National Academy of Sciences of the United States of America. 109 (31): 12509–14. Bibcode:2012PNAS..10912509B. doi:10.1073/pnas.1207892109. PMC 3411975. PMID 22802671.
- Ponte-Sucre A, ed. (2009). ABC Transporters in Microorganisms. Caister Academic. ISBN 978-1-904455-49-3.
- Kamerlin SC, Warshel A (May 2010). "At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis?". Proteins. 78 (6): 1339–75. doi:10.1002/prot.22654. PMC 2841229. PMID 20099310.
- Howard J (2001). Mechanics of motor proteins and the cytoskeleton (1st ed.). Sunderland,MA: Sinauer Associates. ISBN 9780878933334.
- Callaway DJ, Matsui T, Weiss T, Stingaciu LR, Stanley CB, Heller WT, Bu Z (Apr 2017). "Controllable Activation of Nanoscale Dynamics in a Disordered Protein Alters Binding Kinetics". Journal of Molecular Biology. 429 (7): 987–998. doi:10.1016/j.jmb.2017.03.003. PMC 5399307. PMID 28285124.
- Gerstein M, Lesk AM, Chothia C (Jun 1994). "Structural mechanisms for domain movements in proteins". Biochemistry. 33 (22): 6739–49. doi:10.1021/bi00188a001. PMID 8204609.
- Nicholl ID, Matsui T, Weiss TM, Stanley CB, Heller WT, Martel A, Farago B, Callaway DJ, Bu Z (Aug 21, 2018). "Alpha-catenin structure and nanoscale dynamics in solution and in complex with F-actin". Biophysical Journal. 115 (4): 642–654. Bibcode:2018BpJ...115..642N. doi:10.1016/j.bpj.2018.07.005. hdl:2436/621755. PMC 6104293. PMID 30037495.
- Voet D (2011). Biochemistry. Voet, Judith G. (4th ed.). Hoboken, NJ: John Wiley & Sons. ISBN 9780470570951. OCLC 690489261.
- Herzberg O, Chen CC, Kapadia G, McGuire M, Carroll LJ, Noh SJ, Dunaway-Mariano D (Apr 1996). "Swiveling-domain mechanism for enzymatic phosphotransfer between remote reaction sites". Proceedings of the National Academy of Sciences of the United States of America. 93 (7): 2652–7. Bibcode:1996PNAS...93.2652H. doi:10.1073/pnas.93.7.2652. PMC 39685. PMID 8610096.
- Janin J, Wodak SJ (1983). "Structural domains in proteins and their role in the dynamics of protein function". Progress in Biophysics and Molecular Biology. 42 (1): 21–78. doi:10.1016/0079-6107(83)90003-2. PMID 6353481.
- Hayward S (Sep 1999). "Structural principles governing domain motions in proteins". Proteins. 36 (4): 425–35. doi:10.1002/(SICI)1097-0134(19990901)36:4<425::AID-PROT6>3.0.CO;2-S. PMID 10450084. S2CID 29808315.
- Khade, Pranav M.; Kumar, Ambuj; Jernigan, Robert L. (2020-01-17). "Characterizing and Predicting Protein Hinges for Mechanistic Insight". Journal of Molecular Biology. 432 (2): 508–522. doi:10.1016/j.jmb.2019.11.018. ISSN 1089-8638. PMC 7029793. PMID 31786268.
- Khade, Pranav M.; Scaramozzino, Domenico; Kumar, Ambuj; Lacidogna, Giuseppe; Carpinteri, Alberto; Jernigan, Robert L. (2021-11-16). "hdANM: a new comprehensive dynamics model for protein hinges". Biophysical Journal. 120 (22): 4955–4965. doi:10.1016/j.bpj.2021.10.017. ISSN 1542-0086. PMC 8633836. PMID 34687719.
- Meador WE, Means AR, Quiocho FA (Aug 1992). "Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex". Science. 257 (5074): 1251–1255. Bibcode:1992Sci...257.1251M. doi:10.1126/science.1519061. PMID 1519061.
- Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A (May 1992). "Solution structure of a calmodulin-target peptide complex by multidimensional NMR". Science. 256 (5057): 632–638. Bibcode:1992Sci...256..632I. doi:10.1126/science.1585175. PMID 1585175.
- Tokuriki N, Tawfik DS (Apr 2009). "Protein dynamism and evolvability". Science. 324 (5924): 203–207. Bibcode:2009Sci...324..203T. doi:10.1126/science.1169375. PMID 19359577. S2CID 28576496.
- Dyson HJ, Wright PE (Mar 2005). "Intrinsically unstructured proteins and their functions". Nature Reviews Molecular Cell Biology. 6 (3): 197–208. doi:10.1038/nrm1589. PMID 15738986. S2CID 18068406.