DNA re-replication
DNA re-replication (or simply rereplication) is an undesirable and possibly fatal occurrence in eukaryotic cells in which the genome is replicated more than once per cell cycle.[1] Rereplication is believed to lead to genomic instability and has been implicated in the pathologies of a variety of human cancers.[2] To prevent rereplication, eukaryotic cells have evolved multiple, overlapping mechanisms to inhibit chromosomal DNA from being partially or fully rereplicated in a given cell cycle. These control mechanisms rely on cyclin-dependent kinase (CDK) activity.[1] DNA replication control mechanisms cooperate to prevent the relicensing of replication origins and to activate cell cycle and DNA damage checkpoints.[2] DNA rereplication must be strictly regulated to ensure that genomic information is faithfully transmitted through successive generations.
Initiating replication at origins
Replication of DNA always begins at an origin of replication. In yeast, the origins contain autonomously replicating sequences (ARS), distributed throughout the chromosome about 30 kb from each other. They allow replication of DNA wherever they are placed. Each one is 100-200 bp long, and the A element is one of the most conserved stretches. Along with other conserved B elements, they form the section where the ORCs assemble to begin replication. The repetition of these sequences may be the most important to origin recognition.
In animal cells, replication origins may seem to be randomly placed throughout the chromosome, sometimes even acting as ARSs, but local chromatin structure plays a large role in determining where replication will occur. The replication origins are not distributed evenly throughout the chromosome. Replicon clusters, containing 20-80 origins per cluster, are activated at the same time during S phase. Although they are all activated during S phase, heterochromatin tends to be replicated in late S phase, as they are more difficult to access than euchromatin. Epigenetic factors also have a large influence on what gets replicated and when it gets replicated.[3]
Origin licensing
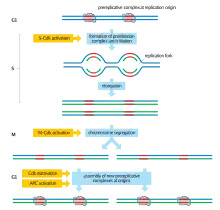
All known mechanisms that prevent DNA rereplication in eukaryotic organisms inhibit origin licensing.[1] Origin licensing is the preliminary step for normal replication initiation during late G1 and early S phase and involves the recruitment of the pre-replicative complex (pre-RC) to the replication origins. Licensing begins with the binding of the multi-subunit ATPase, the origin recognition complex (ORC), to the DNA at the replication origins.[4] Once bound to chromatin the ORC recruits the AAA+ ATPase Cdc6 and the coiled-coil domain protein Cdt1. Cdt1 binding and the ATPase activity of ORC and Cdc6 facilitate the loading of the minichromosome maintenance (MCM) proteins 2-7 onto the chromatin.[1] The MCM complex is the DNA helicase that opens the helix at the replication origin and unwinds the two strands as the replication forks travel along the DNA.[5] Elevated CDK activity at the end of G1 triggers the firing of the origins and the dismantling of the pre-RCs. High CDK levels, which are maintained until the end of mitosis, inhibit or destroy pre-RC components and prevent the origin from relicensing. A new MCM complex cannot be loaded onto the origin until the pre-RC subunits are reactivated with the decline of CDK activity at the end of mitosis. Thus, CDKs serve a dual role in the regulation of eukaryotic DNA replication: elevated CDK activity initiates replication at the origins and prevents rereplication by inhibiting origin re-licensing.[6][7][8] This ensures that no replication origin fires twice in the same cell cycle.[5]
Two-state model for DNA replication regulation
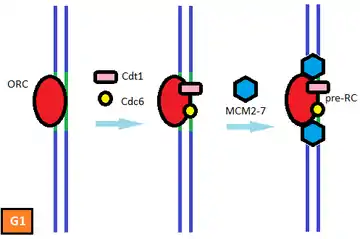
Early experimental evidence on the regulation of DNA replication suggests that replication origins exist in one of two states during the cell cycle: a prereplicative state in G1 and a postreplicative state from the moment of initiation until passage through mitosis.[1] Origins of replication alternate between these two distinct states during the cell cycle.[9] A licensing factor which is required for replication initiation binds to origins in the prereplicative state. At the G1/S transition, the factor is inactivated and cannot be restored until the cell cycle has concluded.[10] The identification and characterization of the ORC, Cdc6, Cdt1, and the MCM complex proteins as the licensing factor gives credence to this model and suggests a means by which the oscillatory nature of CDKs in the cell cycle can regulate rereplication.[1]
Replication regulation
Budding yeast
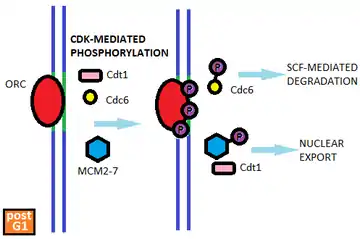
Rereplication regulation is best understood in budding yeast. Saccharomyces cerevisiae cells prevent rereplication by directly regulating pre-RC assembly through the CDK-mediated phosphorylation of the pre-RC components Cdc6, MCM2-7, and the ORC subunits.[5] The phosphorylation of these components is initiated at the onset of S phase and is maintained throughout the rest of the cell cycle as CDK activity remains high. Phosphorylated Cdc6 is bound by the ubiquitin-protein ligase SCF which leads to its proteolytic degradation. CDK-dependent phosphorylation of the MCM2-7 proteins results in the complex's export from the nucleus. (Cdt1 which associates with the MCM complex is similarly exported from the nucleus). Phosphorylation of the ORC subunits presumably disrupts the ORC's ability to bind other pre-RC components.[5] Thus, multiple mechanisms ensure that the pre-RC cannot be reassembled on postreplicative origins.
Note: Since origins fire at different times throughout S phase, it is crucial that the inhibitory mechanisms that prevent new MCM2-7 recruitment do not destabilize existing pre-RCs. Pre-RCs can remain assembled on origins that haven't fired even though rereplication inhibitory mechanisms are inhibiting or destroying pre-RC components.
Other organisms
Although CDK regulation of pre-RC assembly appears to be highly evolutionarily conserved, some differences across organisms are noted. In multicellular eukaryotes pre-RC assembly is regulated by the anaphase-promoting complex (APC) in addition to CDKs. APC, an E3 enzyme, ubiquitinates the protein geminin and targets it for degradation.[5] Geminin normally prevents origin licensing by binding to and inhibiting Cdt1. In G1, APC activity is adequate to suppress the accumulation of geminin, thereby indirectly promoting pre-RC assembly. At the end of G1, APC is inactivated and geminin can accumulate and prevent origin re-licensing.
Cdt1 is usually upregulated by E2F-mediated transcriptional activation and by binding of human acetylase to Orc1. Proteolytic degradation of Cdt1 is a conserved mechanism in various higher order eukaryotes as well. Cdt1 is degraded through the Cul4–Ddb1–Cdt2 E3 ubiquitin ligase complex so that DNA licensing control is maintained in S and G2. Cdt1 is an important regulatory protein, and evolution has led to different pathways of regulation in different organisms. Overexpression of Cdt1 or incactivation of Geminin can lead to re-replication, as undegraded Cdt1 will induce pre-RC assembly.[11]
Pre-RC regulation in most animals is still not well understood.[5]
Consequences of rereplication in eukaryotic cells
Rereplication and mitotic failure are generally not programmed events, but rather result spontaneously from defects in the cell cycle machinery.[1] Rereplication appears to give rise to dsDNA breaks which triggers a DNA damage response and arrests cells in G2.[12] The checkpoint effectively causes a permanent cell cycle arrest and eventual apoptosis.[13]
Rereplication can be experimentally induced by simultaneously disrupting several of the mechanisms that prevent origin re-licensing. For example, deregulation of the ORC, MCM2-7 and Cdc6 mechanisms can induce rereplication in budding yeast cells.[14]
Note: Recent evidence suggests that although overlapping, the multiple replication regulation mechanisms should not be considered as functionally redundant; although a single mechanism may repress rereplication at greater than 99% efficiency, it may not be sufficient to maintain genome stability over many generations.[15] Instead, it is believed that the multiplicative effect of many overlapping mechanisms is what sufficiently prevents rereplication and ensures the faithful transmission of a cell's genome.
Preventing rereplication
Cells with replication stress activate replication checkpoints so that S phase is delayed and slows down the transition to G2/M phase. When replicative stress is recognized by U-2-OS cells, human osteosarcoma cell lines with wild-type retinoblastoma (RB) and p53, the ATM/ATR-regulated DNA damage network is activated.[16] This checkpoint response activates due to overexpression of cyclin E, which has been shown to be important in regulating the licensing system.[17] When cyclin E is overexpressed in U-2-OS cell lines, the ATM/ATR-regulated DNA damage network results in increases in Ser 15-phosphorylated p53, γ-H2AX, and Ser 966-phosphorylated cohesin SMC1.[16] The DNA re-replication response is different from the response taken when damage is due to oxygen radical generation. Damage from oxygen radical generations leads to a response from the Myc oncogene, which phosphorylates p53 and H2AX.[16]
The ATM/ATR DNA damage network will also respond to cases where there is an overexpression of Cdt1. Overexpression of Cdt1 leads to accumulation of ssDNA and DSBs. Ataxia telangiectasia and Rad3 related (ATR) is activated earlier when it detects ssDNA in the earlier phases of DNA re-replication. ATR phosphorylates downstream replication factors, such as RPA2 and MCM2 or through modulation of Rb or p53. Ataxia telangiectasia mutated (ATM) activates after a larger amount of DSBs is detected at later stages of DNA re-replication. While ATM plays a role in cell cycle arrest, apoptosis, and senescence, it is also suspected to play a role in mediating DSB repair, but the exact mechanisms are not understood yet.[11]
Rereplication in cancer
Rereplication has been implicated in tumorigenesis in model organisms and humans. Replication initiation proteins are overexpressed in tissue samples from several types of human cancers[1][18][19] and experimental overexpression of Cdt1 and Cdc6 can cause tumor development in mouse cells.[20][21][22] Similarly, Geminin ablation in knockout mice has been reported to enhance tumor formation.[23] Further, these studies indicate that rereplication can result in an increase in aneuploidy, chromosomal fusions, and DNA breaks.[24] A thorough understanding of the regulatory replication mechanisms is important for the development of novel cancer treatments.
In yeast, increased activity of G1 CDK activity usually inhibits the assembly of pre-RCs and entry into S phase with less active origins, but in cancer cells, p53 and Rb/E2F pathways are deregulated and allow entry into S phase with a reduced amount of active origins. This leads to double-strand breaks in the DNA, increased recombination, and incorrect chromosomal arrangements. The mechanism by which this damage occurs is still not known. One possibility is that reduced origin activation leads to incomplete DNA replication. Significant re-replication is only observed when all CDK regulatory pathways are inhibited.[25]
In mammalian cells, Cdt1 and Cdc6 are much more important to re-replication regulation.[25] Overexpression of Cdt1 and Cdc6 were found in 43/75 cases of non-small cell lung carcinomas.[11] Targeting Cdc6 or ORC in mammalian cells does not cause substantial re-replication. Overexpression of Cdt1, on the other hand, can lead to potentially lethal re-replication levels on its own. This response is seen only in cancer cells.[25] Overexpression of E2F family members contributes to an increase in Cdt1 and Cdc6 expression. Loss of p53 regulation in cells can also be observed frequently in cell lines that overexpress Cdt1 or Cdc6.[11]
Endoreduplication
For the special case of cell cycle-regulated DNA replication in which DNA synthesis is uncoupled from cell cycle progression refer to endoreduplication. Endoreduplication is an important and widespread mechanism in many cell types. It does not adhere to many of the cell cycle checkpoints and damage controls in regularly dividing cells, but it does not result in uncontrolled re-replication. Endoreduplication is a controlled process and occurs to perform a specific cell function. In some cells, it has been proposed that endoreduplication is used as a way to store nucleotides for embryogenesis and germination. In other cases, endoreduplication may be used in cells that are only used for storage of nutrients. Despite its useful functioning in many cells, endoreduplication has also been observed in cancerous cells, and it is not fully understood whether endoreduplication leads to cancerous behavior or whether other mutations lead to endoreduplication. Other mechanisms may be involved in mediating these changes.[26]
References
- Arias EE, Walter JC (March 2007). "Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells". Genes & Development. 21 (5): 497–518. doi:10.1101/gad.1508907. PMID 17344412.
- Truong LN, Wu X (February 2011). "Prevention of DNA re-replication in eukaryotic cells". Journal of Molecular Cell Biology. 3 (1): 13–22. doi:10.1093/jmcb/mjq052. PMC 3030972. PMID 21278447.
- Morgan, D. O. (2007). The cell cycle: principles of control. New Science Press.
- Cvetic CA, Walter JC (January 2006). "Getting a grip on licensing: mechanism of stable Mcm2-7 loading onto replication origins". Molecular Cell. 21 (2): 143–4. doi:10.1016/j.molcel.2006.01.003. PMID 16427002.
- Morgan, David O. (2007). The Cell Cycle: Principles of Control. Oxford University Press. ISBN 978-0-87893-508-6.
- Bell SP, Dutta A (2002). "DNA replication in eukaryotic cells". Annual Review of Biochemistry. 71: 333–74. doi:10.1146/annurev.biochem.71.110601.135425. PMID 12045100.
- Broek D, Bartlett R, Crawford K, Nurse P (January 1991). "Involvement of p34cdc2 in establishing the dependency of S phase on mitosis". Nature. 349 (6308): 388–93. Bibcode:1991Natur.349..388B. doi:10.1038/349388a0. PMID 1992340. S2CID 4263847.
- Hayles J, Fisher D, Woollard A, Nurse P (September 1994). "Temporal order of S phase and mitosis in fission yeast is determined by the state of the p34cdc2-mitotic B cyclin complex". Cell. 78 (5): 813–22. doi:10.1016/S0092-8674(94)90542-8. PMID 8087848. S2CID 7449103.
- Diffley JF, Cocker JH, Dowell SJ, Rowley A (July 1994). "Two steps in the assembly of complexes at yeast replication origins in vivo". Cell. 78 (2): 303–16. doi:10.1016/0092-8674(94)90299-2. PMID 8044842. S2CID 44884064.
- Blow JJ, Laskey RA (April 1988). "A role for the nuclear envelope in controlling DNA replication within the cell cycle". Nature. 332 (6164): 546–8. Bibcode:1988Natur.332..546B. doi:10.1038/332546a0. PMID 3357511. S2CID 4313693.
- Lan N. Truong, Xiaohua Wu; Prevention of DNA re-replication in eukaryotic cells, Journal of Molecular Cell Biology, Volume 3, Issue 1, 1 February 2011, Pages 13–22
- Green BM, Li JJ (January 2005). "Loss of rereplication control in Saccharomyces cerevisiae results in extensive DNA damage". Molecular Biology of the Cell. 16 (1): 421–32. doi:10.1091/mbc.E04-09-0833. PMC 539184. PMID 15537702.
- Archambault V, Ikui AE, Drapkin BJ, Cross FR (August 2005). "Disruption of mechanisms that prevent rereplication triggers a DNA damage response". Molecular and Cellular Biology. 25 (15): 6707–21. doi:10.1128/MCB.25.15.6707-6721.2005. PMC 1190345. PMID 16024805.
- Nguyen VQ, Co C, Li JJ (June 2001). "Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms". Nature. 411 (6841): 1068–73. Bibcode:2001Natur.411.1068N. doi:10.1038/35082600. PMID 11429609. S2CID 4393812.
- Green BM, Morreale RJ, Ozaydin B, Derisi JL, Li JJ (May 2006). "Genome-wide mapping of DNA synthesis in Saccharomyces cerevisiae reveals that mechanisms preventing reinitiation of DNA replication are not redundant". Molecular Biology of the Cell. 17 (5): 2401–14. doi:10.1091/mbc.E05-11-1043. PMC 1446083. PMID 16481397.
- Bartkova, J., Hořejší, Z., Koed, K., Krämer, A., Tort, F., Zieger, K., ... & Ørntoft, T. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature, 434(7035), 864.
- Blow, J. J., & Dutta, A. (2005). Preventing re-replication of chromosomal DNA. Nature reviews Molecular cell biology, 6(6), 476.
- Blow JJ, Gillespie PJ (October 2008). "Replication licensing and cancer--a fatal entanglement?". Nature Reviews. Cancer. 8 (10): 799–806. doi:10.1038/nrc2500. PMC 2577763. PMID 18756287.
- Gonzalez MA, Tachibana KE, Laskey RA, Coleman N (February 2005). "Control of DNA replication and its potential clinical exploitation". Nature Reviews. Cancer. 5 (2): 135–41. doi:10.1038/nrc1548. PMID 15660109. S2CID 22492421.
- Arentson E, Faloon P, Seo J, Moon E, Studts JM, Fremont DH, Choi K (February 2002). "Oncogenic potential of the DNA replication licensing protein CDT1". Oncogene. 21 (8): 1150–8. doi:10.1038/sj.onc.1205175. PMID 11850834.
- Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, Kotsinas A, Nahum O, Zoumpourlis V, Kouloukoussa M, Lygerou Z, Taraviras S, Kittas C, Bartkova J, Papavassiliou AG, Bartek J, Halazonetis TD, Gorgoulis VG (November 2007). "Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior". Cancer Research. 67 (22): 10899–909. doi:10.1158/0008-5472.CAN-07-2837. PMID 18006835.
- Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, Choi K (December 2005). "Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53". Oncogene. 24 (55): 8176–86. doi:10.1038/sj.onc.1208881. PMID 16261166.
- Champeris Tsaniras S, Villiou M, Giannou AD, Nikou S, Petropoulos M, Pateras IS, Tserou P, Karousi F, Lalioti ME, Gorgoulis VG, Patmanidi AL, Stathopoulos GT, Bravou V, Lygerou Z, Taraviras S (2018). "Geminin ablation in vivo enhances tumorigenesis through increased genomic instability". Journal of Pathology. 246 (2): 134–140. doi:10.1002/path.5128. PMID 29952003. S2CID 49474213.
- Hook SS, Lin JJ, Dutta A (December 2007). "Mechanisms to control rereplication and implications for cancer". Current Opinion in Cell Biology. 19 (6): 663–71. doi:10.1016/j.ceb.2007.10.007. PMC 2174913. PMID 18053699.
- Hills, S. A., & Diffley, J. F. (2014). DNA replication and oncogene-induced replicative stress. Current biology, 24(10), R435-R444
- Lee, H. O., Davidson, J. M., & Duronio, R. J. (2009). Endoreplication: polyploidy with purpose. Genes & development, 23(21), 2461-2477.