Severe achondroplasia with developmental delay and acanthosis nigricans
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) is a very rare genetic disorder. This disorder is one that affects bone growth and is characterized by skeletal, brain, and skin abnormalities.[3][4] Those affected by the disorder are severely short in height and commonly possess shorter arms and legs.[3][4] In addition, the bones of the legs are often bowed and the affected have smaller chests with shorter rib bones, along with curved collarbones.[3][4] Other symptoms of the disorder include broad fingers and extra folds of skin on the arms and legs.[3][4] Developmentally, many individuals who suffer from the disorder show a higher level in delays and disability. Seizures are also common due to structural abnormalities of the brain.[3][4] Those affected may also suffer with apnea, the slowing or loss of breath for short periods of time.[3][4]
| Severe achondroplasia with developmental delay and acanthosis nigricans | |
|---|---|
| Other names | SADDAN[1] |
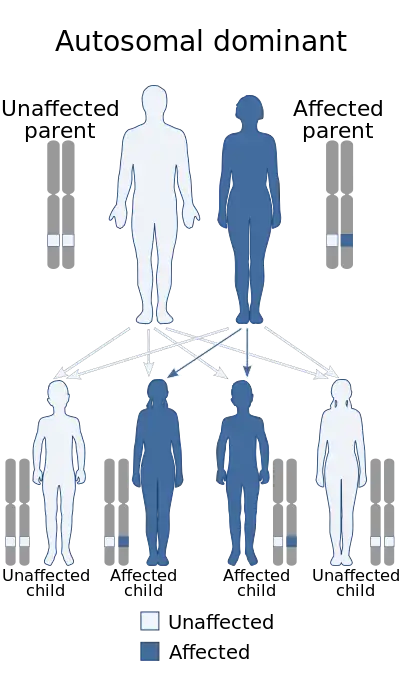 | |
| Severe achondroplasia with developmental delay and acanthosis nigricans is inheried in an autosomal dominant manner.[2] | |
Many of the features of SADDAN are similar to those seen in other skeletal disorders, specifically achondroplasia and thanatophoric dysplasia. Achondroplasia is a form of short-limbed dwarfism. This type of dwarfism is caused by the inability of the cartilage of the skeleton to ossify and turn to bone.[5] Acanthosis nigricans is a skin condition in which areas of the skin is of a dark and velvety discoloration, often seen in the body folds and creases such as the armpits, groin, and neck.[6] Within those affected by SADDAN, acanthosis nigricans develops early on, usually in infancy or early childhood.[4]
Genetic
The mutated gene responsible for the disorder is the FGFR3 gene, more specifically; a Lys650Met missense mutation of the FGFR3 gene is what causes SADDAN.[7] This gene codes for the instructions of a protein that is integral in the development and maintenance of bone and brain tissue.[4] Mutations of this gene cause the protein to be overly active, causing many characteristics of this disorder.[4]
SADDAN is an autosomal dominant genetic disorder.[4] Autosomal means that the gene responsible for the mutation and disorder is found on a non-sex chromosome and that either the mother or father can pass on the gene, while dominant means that only one copy of the gene is required for the individual to have the disorder.
The disorder is very rare and has only been described in a few number of cases worldwide.[4] While the disorder can be genetically inherited, no instances of inheritance have been recorded as of yet. Rather, of the few cases documented, the individual affected by the disorder is affected as a product of a random mutation, also called a de novo mutation, of the FGFR3 gene only, not by inheritance of the mutated gene.[4]
Diagnosis
Medical diagnosis is required. Clinical tests can be performed, as well as molecular genetic testing. The available tests include:
Sequence analysis of the entire coding region[3]
- Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) - Sanger Sequencing: Diagnosis, Mutation Confirmation, Pre-symptomatic, Risk Assessment, Screening
- Craniosynostosis: Diagnosis
- Invitae FGFR3-Related Disorders Test: Pre-symptomatic, Diagnosis, Therapeutic management
Mutation scanning of select exons[3]
- Skeletal Dysplasia Panel: Diagnosis, Prognostic
Sequence analysis of select exons[3]
- Severe Achondroplasia with Developmental Delay and Acanthosis Nigricans (SADDAN, FGFR3): Diagnosis, Mutation Confirmation, Risk Assessment
- Severe Achondroplasia, Developmental Delay, Acanthosis Nigricans: Diagnosis, Mutation Confirmation
Deletion/duplication analysis[3]
- Invitae FGFR3-Related Disorders Test: Pre-symptomatic, Diagnosis, Therapeutic management
Management
Life with SADDAN is manageable, although therapy, surgery, and lifelong doctor surveillance may be required.[7]
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Severe achondroplasia developmental delay acanthosis nigricans syndrome". www.orpha.net. Retrieved 24 October 2019.
- "OMIM Entry - # 616482 - ACHONDROPLASIA, SEVERE, WITH DEVELOPMENTAL DELAY AND ACANTHOSIS NIGRICANS; SADDAN". omim.org. Retrieved 6 January 2018.
- National Center for Biotechnology Information. Severe achondroplasia with developmental delay and acanthosis nigricans. Retrieved from https://www.ncbi.nlm.nih.gov/gtr/conditions/C2674173/
- U.S. National Library of Medicine. (2012). Saddan. Retrieved from https://ghr.nlm.nih.gov/condition/saddan#sourcesforpage
- U.S. National Library of Medicine. (2012). Achondroplasia. Retrieved from https://ghr.nlm.nih.gov/condition/achondroplasia#
- Mayo Clinic Staff. (2015). Acanthosis nigricans. Retrieved from http://www.mayoclinic.org/diseases-conditions/acanthosis-nigricans/basics/definition/con-20025600
- Pauli, R. M. (2012). Achondroplasia. GeneReviews (). Seattle, Washington: Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1152/
- Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, Jabs EW, Curry CJ, Wilcox WR, Lachman RS, Rimoin DL, Francomano CA (1999). "Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3". Am J Med Genet. 85 (1): 53–65. doi:10.1002/(SICI)1096-8628(19990702)85:1<53::AID-AJMG10>3.0.CO;2-F. PMID 10377013.
- Cohen MM Jr (2002). "Some chondrodysplasias with short limbs: molecular perspectives". Am J Med Genet. 112 (3): 304–13. doi:10.1002/ajmg.10780. PMID 12357475.
- Vajo Z, Francomano CA, Wilkin DJ (2000). "The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans". Endocr Rev. 21 (1): 23–39. doi:10.1210/edrv.21.1.0387. PMID 10696568.