SN2 reaction
The SN2 reaction is a type of reaction mechanism that is common in organic chemistry. In this mechanism, one bond is broken and one bond is formed in a concerted way, i.e., in one step. The name SN2 refers to the Hughes-Ingold symbol of the mechanism: "SN" indicates that the reaction is a nucleophilic substitution, and "2" that it proceeds via a bi-molecular mechanism, which means both the reacting species are involved in the rate-determining step. The other major type of nucleophilic substitution is the SN1,[1] but many other more specialized mechanisms describe substitution reactions.
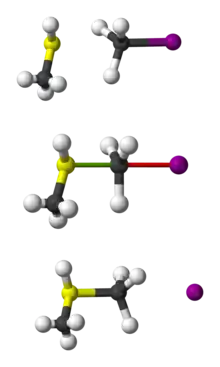
The SN2 reaction can be considered as an analogue of the associative substitution in the field of inorganic chemistry.
Reaction mechanism
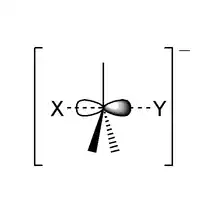
The reaction most often occurs at an aliphatic sp3 carbon center with an electronegative, stable leaving group attached to it (often denoted X), which is frequently a halide atom. The breaking of the C–X bond and the formation of the new bond (often denoted C–Y or C–Nu) occur simultaneously through a transition state in which a carbon under nucleophilic attack is pentacoordinate, and approximately sp2 hybridised. The nucleophile attacks the carbon at 180° to the leaving group, since this provides the best overlap between the nucleophile's lone pair and the C–X σ* antibonding orbital. The leaving group is then pushed off the opposite side and the product is formed with inversion of the tetrahedral geometry at the central atom.
If the substrate under nucleophilic attack is chiral, then this often leads to inversion of configuration (stereochemistry), called a Walden inversion.
In an example of the SN2 reaction, the attack of Br− (the nucleophile) on an ethyl chloride (the electrophile) results in ethyl bromide, with chloride ejected as the leaving group.
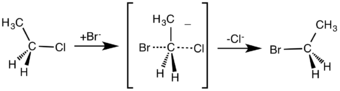 SN2 reaction of chloroethane with bromide ion
SN2 reaction of chloroethane with bromide ion- If the molecule that is undergoing SN2 reaction has a chiral centre, then it is possible that the optical activity of the product would be different from that of the reactant. In an example, 1-bromo-1-fluoroethane can undergo SN2 reaction to form 1-fluoroethan-1-ol, with the nucleophile being an OH− group. In this case, if the reactant is levorotatory, then the product would be dextrorotatory, and vice versa.[2]
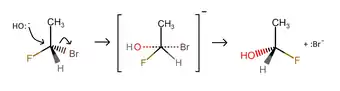 SN2 mechanism of 1-bromo-1-fluoroethane with one of the carbon atoms being a chiral centre.
SN2 mechanism of 1-bromo-1-fluoroethane with one of the carbon atoms being a chiral centre.
SN2 attack occurs if the backside route of attack is not sterically hindered by substituents on the substrate (ethyl chloride being the substrate above). Therefore, this mechanism usually occurs at unhindered primary and secondary carbon centres. If there is steric crowding on the substrate near the leaving group, such as at a tertiary carbon centre, the substitution will involve an SN1 rather than an SN2 mechanism, (an SN1 would also be more likely in this case because a sufficiently stable carbocation intermediary could be formed).
Factors affecting the rate of the reaction
The four factors that affect the rate of the reaction, in the order of decreasing importance, are:[3][4]
Substrate
The substrate plays the most important part in determining the rate of the reaction. This is because the nucleophile attacks from the back of the substrate, thus breaking the carbon-leaving group bond and forming the carbon-nucleophile bond. Therefore, to maximise the rate of the SN2 reaction, the back of the substrate must be as unhindered as possible. Overall, this means that methyl and primary substrates react the fastest, followed by secondary substrates. Tertiary substrates do not participate in SN2 reactions, because of steric hindrance. Structures that can form highly stable cations by simple loss of the leaving group, for example, as a resonance-stabilized carbocation, are especially likely to react via an SN1 pathway in competition with SN2.
Nucleophile
Like the substrate, steric hindrance affects the nucleophile's strength. The methoxide anion, for example, is both a strong base and nucleophile because it is a methyl nucleophile, and is thus very much unhindered. tert-Butoxide, on the other hand, is a strong base, but a poor nucleophile, because of its three methyl groups hindering its approach to the carbon. Nucleophile strength is also affected by charge and electronegativity: nucleophilicity increases with increasing negative charge and decreasing electronegativity. For example, OH− is a better nucleophile than water, and I− is a better nucleophile than Br− (in polar protic solvents). In a polar aprotic solvent, nucleophilicity increases up a column of the periodic table as there is no hydrogen bonding between the solvent and nucleophile; in this case nucleophilicity mirrors basicity. I− would therefore be a weaker nucleophile than Br− because it is a weaker base. Verdict - A strong/anionic nucleophile always favours SN2 manner of nucleophillic substitution.
Leaving group
The stability of the leaving group as an anion and the strength of its bond to the carbon atom both affect the rate of reaction. The more stable the conjugate base of the leaving group is, the more likely that it will take the two electrons of its bond to carbon during the reaction. Therefore, the weaker the leaving group is as a conjugate base, and thus the stronger its corresponding acid, the better the leaving group. Examples of good leaving groups are therefore the halides (except fluoride, due to its strong bond to the carbon atom) and tosylate, whereas HO− and H2N− are not.
Solvent
The solvent affects the rate of reaction because solvents may or may not surround a nucleophile, thus hindering or not hindering its approach to the carbon atom.[5] Polar aprotic solvents, like tetrahydrofuran, are better solvents for this reaction than polar protic solvents because polar protic solvents will hydrogen bond to the nucleophile, hindering it from attacking the carbon with the leaving group. A polar aprotic solvent with low dielectric constant or a hindered dipole end will favour SN2 manner of nucleophilic substitution reaction. Examples: dimethylsulfoxide, dimethylformamide, acetone, etc. In parallel, solvation also has a significant impact on the intrinsic strength of the nucleophile, in which strong interactions between solvent and the nucleophile, found for polar protic solvents, furnish a weaker nucleophile. In contrast, polar aprotic solvents can only weakly interact with the nucleophile, and thus, are to a lesser extent able to reduce the strength of the nucleophile.[6][7]
Reaction kinetics
The rate of an SN2 reaction is second order, as the rate-determining step depends on the nucleophile concentration, [Nu−] as well as the concentration of substrate, [RX].[8]
- r = k[RX][Nu−]
This is a key difference between the SN1 and SN2 mechanisms. In the SN1 reaction the nucleophile attacks after the rate-limiting step is over, whereas in SN2 the nucleophile forces off the leaving group in the limiting step. In other words, the rate of SN1 reactions depend only on the concentration of the substrate while the SN2 reaction rate depends on the concentration of both the substrate and nucleophile.[8]
It has been shown[9] that except in uncommon (but predictable cases) primary and secondary substrates go exclusively by the SN2 mechanism while tertiary substrates go via the SN1 reaction. There are two factors which complicate determining the mechanism of nucleophilic substitution reactions at secondary carbons:
- Many reactions studied are solvolysis reactions where a solvent molecule (often an alcohol) is the nucleophile. While still a second order reaction mechanistically, the reaction is kinetically first order as the concentration of the nucleophile–the solvent molecule, is effectively constant during the reaction. This type of reaction is often called a pseudo first order reaction.
- In reactions where the leaving group is also a good nucleophile (bromide for instance) the leaving group can perform an SN2 reaction on a substrate molecule. If the substrate is chiral, this inverts the configuration of the substrate before solvolysis, leading to a racemized product–the product that would be expected from an SN1 mechanism. In the case of a bromide leaving group in alcoholic solvent Cowdrey et al.[10] have shown that bromide can have an SN2 rate constant 100-250 times higher than the rate constant for ethanol. Thus, after only a few percent solvolysis of an enantiospecific substrate, it becomes racemic.
The examples in textbooks of secondary substrates going by the SN1 mechanism invariably involve the use of bromide (or other good nucleophile) as the leaving group have confused the understanding of alkyl nucleophilic substitution reactions at secondary carbons for 80 years[3]. Work with the 2-adamantyl system (SN2 not possible) by Schleyer and co-workers,[11] the use of azide (an excellent nucleophile but very poor leaving group) by Weiner and Sneen,[12][13] the development of sulfonate leaving groups (non-nucleophilic good leaving groups), and the demonstration of significant experimental problems in the initial claim of an SN1 mechanism in the solvolysis of optically active 2-bromooctane by Hughes et al.[14][3] have demonstrated conclusively that secondary substrates go exclusively (except in unusual but predictable cases) by the SN2 mechanism.
E2 competition
A common side reaction taking place with SN2 reactions is E2 elimination: the incoming anion can act as a base rather than as a nucleophile, abstracting a proton and leading to formation of the alkene. This pathway is favored with sterically hindered nucleophiles. Elimination reactions are usually favoured at elevated temperatures[15] because of increased entropy. This effect can be demonstrated in the gas-phase reaction between a sulfonate and a simple alkyl bromide taking place inside a mass spectrometer:[16][17]
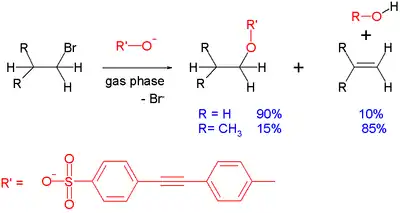
With ethyl bromide, the reaction product is predominantly the substitution product. As steric hindrance around the electrophilic center increases, as with isobutyl bromide, substitution is disfavored and elimination is the predominant reaction. Other factors favoring elimination are the strength of the base. With the less basic benzoate substrate, isopropyl bromide reacts with 55% substitution. In general, gas phase reactions and solution phase reactions of this type follow the same trends, even though in the first, solvent effects are eliminated.
Roundabout mechanism
A development attracting attention in 2008 concerns a SN2 roundabout mechanism observed in a gas-phase reaction between chloride ions and methyl iodide with a special technique called crossed molecular beam imaging. When the chloride ions have sufficient velocity, the initial collision of it with the methyl iodide molecule causes the methyl iodide to spin around once before the actual SN2 displacement mechanism takes place.[18][19][20]
See also
References
- McMurry, John E. (1992), Organic Chemistry (3rd ed.), Belmont: Wadsworth, ISBN 0-534-16218-5
- CURTIS, CLIFF. MURGATROYD, JASON. SCOTT, DAVE (2019). Edexcel international a level chemistry student book. [Place of publication not identified]: EDEXCEL Limited. ISBN 978-1-292-24472-3. OCLC 1084791738.
{{cite book}}: CS1 maint: multiple names: authors list (link) - Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- Hamlin, Trevor A.; Swart, Marcel; Bickelhaupt, F. Matthias (2018). "Nucleophilic Substitution (SN2): Dependence on Nucleophile, Leaving Group, Central Atom, Substituents, and Solvent". ChemPhysChem. 19 (11): 1315–1330. doi:10.1002/cphc.201701363. ISSN 1439-7641. PMC 6001448. PMID 29542853.
- Hamlin, Trevor A.; van Beek, Bas; Wolters, Lando P.; Bickelhaupt, F. Matthias (2018). "Nucleophilic Substitution in Solution: Activation Strain Analysis of Weak and Strong Solvent Effects". Chemistry – A European Journal. 24 (22): 5927–5938. doi:10.1002/chem.201706075. ISSN 1521-3765. PMC 5947303. PMID 29457865.
- Hansen, Thomas; Roozee, Jasper C.; Bickelhaupt, F. Matthias; Hamlin, Trevor A. (4 February 2022). "How Solvation Influences the S N 2 versus E2 Competition". The Journal of Organic Chemistry. 87 (3): 1805–1813. doi:10.1021/acs.joc.1c02354. PMC 8822482. PMID 34932346.
- Vermeeren, Pascal; Hansen, Thomas; Jansen, Paul; Swart, Marcel; Hamlin, Trevor A.; Bickelhaupt, F. Matthias (December 2020). "A Unified Framework for Understanding Nucleophilicity and Protophilicity in the S N 2/E2 Competition". Chemistry – A European Journal. 26 (67): 15538–15548. doi:10.1002/chem.202003831. PMC 7756690. PMID 32866336.
- Clayden, Jonathan; Greeves, Nick; Warren, Stuart (2012). Organic chemistry (2nd ed.). Oxford: Oxford University Press. p. 330. ISBN 978-0-19-927029-3.
- Absence of SN1 Involvement in the Solvolysis of Secondary Alkyl Compounds, T. J. Murphy, J. Chem. Educ.; 2009; 86(4) pp 519-24; (Article) doi: 10.1021/ed041p678
- W.A. Cowdrey; E.D. Hughes; C.K. Ingold; S. Masterman; A.D. Scott (1937). "Relation of Steric orientation to Mechanism in Substitution Involving Halogen Atoms and Simple or Substituted Hydroxyl Groups". J. Chem. Soc.: 1252–1271. doi:10.1039/JR9370001252.
- The 2-Adamantyl System, a Standard for Limiting Solvolysis in a Secondary Substrate J. L. Fry, C. J. Lancelot, L. K. M. Lam, J. M Harris, R. C. Bingham, D. J. Raber, R. E. Hill, P. v. R. Schleyer, J. Am. Chem. Soc.,; 1970; 92, pp 1240-42 (Article); doi: 10.1021/ja00478a031
- A Clarification of the Mechanism of Solvolysis of 2-Octyl Sulfonates. Stereochemical Considerations; H. Weiner, R. A. Sneen, J. Am. Chem. Soc.,; 1965; 87 pp 287-91; (Article) doi: 10.1021/ja01080a026
- A Clarification of the Mechanism of Solvolysis of 2-Octyl Sulfonates. Kinetic Considerations; H. Weiner, R. A. Sneen, J. Am. Chem. Soc.; 1965; 87 pp 292-96; (Article) doi: 10.1021/ja01080a027
- Homogeneous Hydrolysis and Alcoholysis of β-n-Octyl halides, E. D. Hughes, C. K. Ingold, S. Masterman, J. Chem. Soc.; 1937; pp 1196–1201; (Article) doi: 10.1039/JR9370001196
- "Elimination Reactions Are Favored By Heat — Master Organic Chemistry". www.masterorganicchemistry.com. Retrieved 13 April 2018.
- Gas Phase Studies of the Competition between Substitution and Elimination Reactions Scott Gronert Accounts of Chemical Research; 2003; 36(11) pp 848 - 857; (Article) doi:10.1021/ar020042n
- The technique used is electrospray ionization and because it requires charged reaction products for detection the nucleophile is fitted with an additional sulfonate anionic group, non-reactive and well separated from the other anion. The product ratio of substitution and elimination product can be measured from the intensity their relative molecular ions.
- Imaging Nucleophilic Substitution Dynamics J. Mikosch, S. Trippel, C. Eichhorn, R. Otto, U. Lourderaj, J. X. Zhang, W. L. Hase, M. Weidemüller, and R. Wester Science 11 January 2008 319: 183-186 doi:10.1126/science.1150238 (in Reports)
- PERSPECTIVES CHEMISTRY: Not So Simple John I. Brauman (11 January 2008) Science 319 (5860), 168. doi:10.1126/science.1152387
- Surprise From SN2 Snapshots Ion velocity measurements unveil additional unforeseen mechanism Carmen Drahl Chemical & Engineering News January 14, 2008 Volume 86, Number 2 p. 9 http://pubsapp.acs.org/cen/news/86/i02/8602notw1.html, video included