Scaffolding (bioinformatics)
Scaffolding is a technique used in bioinformatics. It is defined as follows:[1]
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
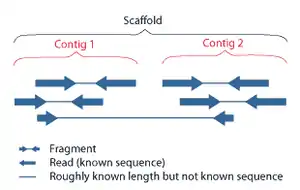
When creating a draft genome, individual reads of DNA are second assembled into contigs, which, by the nature of their assembly, have gaps between them. The next step is to then bridge the gaps between these contigs to create a scaffold.[2] This can be done using either optical mapping or mate-pair sequencing.[3]
Assembly software
The sequencing of the Haemophilus influenzae genome marked the advent of scaffolding. That project generated a total of 140 contigs, which were oriented and linked using paired end reads. The success of this strategy prompted the creation of the software, Grouper, which was included in genome assemblers. Until 2001, this was the only scaffolding software. After the Human Genome Project and Celera proved that it was possible to create a large draft genome, several other similar programs were created. Bambus was created in 2003 and was a rewrite of the original grouper software, but afforded researchers the ability to adjust scaffolding parameters. This software also allowed for optional use of other linking data, such as contig order in a reference genome.[4]
Algorithms used by assembly software are very diverse, and can be classified as based on iterative marker ordering, or graph based. Graph based applications have the capacity to order and orient over 10,000 markers, compared to the maximum 3000 markers capable of iterative marker applications.[5] Algorithms can be further classified as greedy, non greedy, conservative, or non conservative. Bambus uses a greedy algorithm, defined as such because it joins together contigs with the most links first. The algorithm used by Bambus 2 removes repetitive contigs before orienting and ordering them into scaffolds. SSPACE also uses a greedy algorithm that begins building its first scaffold with the longest contig provided by the sequence data. SSPACE is the most commonly cited assembly tool in biology publications, likely due to the fact that it is rated as a significantly more intuitive program to install and run than other assemblers.[6]
In recent years, there has been an advent of new kinds of assemblers capable of integrating linkage data from multiple types of linkage maps. ALLMAPS is the first of such programs and is capable of combining data from genetic maps, created using SNPs or recombination data, with physical maps such as optical or synteny maps.[7]
Some software, like ABySS and SOAPdenovo, contain gap filling algorithms which, although they do not create any new scaffolds, serve to decrease the gap length between contigs of individual scaffolds. A standalone program, GapFiller, is capable of closing a larger amount of gaps, using less memory than gap filling algorithms contained within assembly programs.[8]
Utturkar et al. investigated the utility of several different assembly software packages in combination with hybrid sequence data. They concluded that the ALLPATHS-LG and SPAdes algorithms were superior to other assemblers in terms of the number of, maximum length of, and N50 length of contigs and scaffolds.[9]
Scaffolding and next generation sequencing
Most high-throughput, next generation sequencing platforms produce shorter read lengths compared to Sanger sequencing. These new platforms are able to generate large quantities of data in short periods of time, but until methods were developed for de novo assembly of large genomes from short read sequences, Sanger sequencing remained the standard method of creating a reference genome.[10] Although Illumina platforms are now able to generate mate pair reads with average lengths of 150bp, they were originally only able to generate reads of 75bp or less, which caused many people in the science community to doubt a reliable reference genome could ever be constructed with short read technology. The increased difficulty of contig and scaffold assembly associated with the new technologies has created a demand for powerful new computer programs and algorithms capable of making sense of the data.[11]
One strategy that incorporates high-throughput next generation sequencing is hybrid sequencing, wherein several sequencing technologies are used at different levels of coverage, so that they can complement each other with their respective strengths. The release of the SMRT platform, from Pacific Biosciences, marked the beginning of single molecule sequencing and long read tech. It has been shown that 80-100X coverage with SMRT technology, which generates average read with lengths of 5456bp, is usually sufficient to create a finished de novo assembly for prokaryotic organisms. When the funds for that level of coverage are not available to a researcher, they might decide to use a hybrid approach.
Goldberg et al. evaluated the effectiveness of combining high throughput pyrosequencing with traditional Sanger sequencing. They were able to greatly increase N50 contig length and decrease gap length, and even to close one microbial genome with this approach.[12]
Optical mapping
It has been shown that integration of linkage maps can aid de novo assemblies with long range, chromosome scale recombination data, without which, assemblies can be subject to macro ordering errors. Optical mapping is the process of immobilizing the DNA on a slide and digesting it with restriction enzymes. The fragment ends are then fluorescently tagged and stitched back together. For the last two decades, optical mapping has been prohibitively expensive, but recent advances in technology have reduced cost significantly.[5][13]
See also
- "EDAM Ontology of Bioinformatics Operations and Data Formats".
- Waterston, Robert (2002). "On the Sequencing of the Human Genome". Proceedings of the National Academy of Sciences of the United States of America. 99 (6): 3712–3716. Bibcode:2002PNAS...99.3712W. doi:10.1073/pnas.042692499. PMC 122589. PMID 11880605.
- Flot, Jean-François; Marie-Nelly, Hervé; Koszul, Romain (2015-10-07). "Contact genomics: scaffolding and phasing (meta)genomes using chromosome 3D physical signatures". FEBS Letters. 589 (20 Pt A): 2966–2974. doi:10.1016/j.febslet.2015.04.034. ISSN 1873-3468. PMID 25935414.
- Pop, Mihai; Kosack, Daniel S.; Salzberg, Steven L. (2004-01-01). "Hierarchical Scaffolding With Bambus". Genome Research. 14 (1): 149–159. doi:10.1101/gr.1536204. ISSN 1088-9051. PMC 314292. PMID 14707177.
- Fierst JL (2015) Using linkage maps to correct and scaffold de novo genome assemblies: methods, challenges, and computational tools. In: Frontiers in Genetics. http://journal.frontiersin.org/article/10.3389/fgene.2015.00220/full . Accessed 7 Apr 2017
- Hunt, M; Newbold, C; Berriman, M; Otto, TD (2014). "A comprehensive evaluation of assembly scaffolding tools". Genome Biology. 15 (3): R42. doi:10.1186/gb-2014-15-3-r42. PMC 4053845. PMID 24581555.
- Tang, H; Zhang, X; Miao, C; et al. (2015). "ALLMAPS: robust scaffold ordering based on multiple maps". Genome Biology. 16: 3. doi:10.1186/s13059-014-0573-1. PMC 4305236. PMID 25583564.
- Boetzer, M; Pirovano, W (2012). "Toward almost closed genomes with GapFiller". Genome Biology. 13 (6): R56. doi:10.1186/gb-2012-13-6-r56. PMC 3446322. PMID 22731987.
- Utturkar, SM; Klingeman, DM; Land, ML; et al. (2014). "Evaluation and validation of de novo and hybrid assembly techniques to derive high-quality genome sequences". Bioinformatics. 30 (19): 2709–2716. doi:10.1093/bioinformatics/btu391. PMC 4173024. PMID 24930142.
- Li, Ruiqiang; Zhu, Hongmei; Ruan, Jue; Qian, Wubin; Fang, Xiaodong; Shi, Zhongbin; Li, Yingrui; Li, Shengting; Shan, Gao (2017-02-09). "De novo assembly of human genomes with massively parallel short read sequencing". Genome Research. 20 (2): 265–272. doi:10.1101/gr.097261.109. ISSN 1088-9051. PMC 2813482. PMID 20019144.
- Pareek, Chandra Shekhar; Smoczynski, Rafal; Tretyn, Andrzej (2017-02-09). "Sequencing technologies and genome sequencing". Journal of Applied Genetics. 52 (4): 413–435. doi:10.1007/s13353-011-0057-x. ISSN 1234-1983. PMC 3189340. PMID 21698376.
- Goldberg, Susanne M. D.; et al. (2006). "A Sanger/Pyrosequencing Hybrid Approach for the Generation of High-Quality Draft Assemblies of Marine Microbial Genomes". Proceedings of the National Academy of Sciences of the United States of America. 103 (30): 11240–11245. Bibcode:2006PNAS..10311240G. doi:10.1073/pnas.0604351103. JSTOR 30049789. PMC 1544072. PMID 16840556.
- Chaisson, Mark; Wilson, Richard; Eichler, Evan (7 October 2015). "Genetic variation and the de novo assembly of human genomes". Nature Reviews Genetics. 16 (11): 627–640. doi:10.1038/nrg3933. PMC 4745987. PMID 26442640.