Cellular senescence
Cellular senescence is a phenomenon characterized by the cessation of cell division. In their experiments during the early 1960s, Leonard Hayflick and Paul Moorhead found that normal human fetal fibroblasts in culture reach a maximum of approximately 50 cell population doublings before becoming senescent.[1][2][3] This process is known as "replicative senescence", or the Hayflick limit. Hayflick's discovery of mortal cells paved the path for the discovery and understanding of cellular aging molecular pathways.[4] Cellular senescence can be initiated by a wide variety of stress inducing factors. These stress factors include both environmental and internal damaging events, abnormal cellular growth, oxidative stress, autophagy factors, among many other things.[5]
.svg.png.webp)
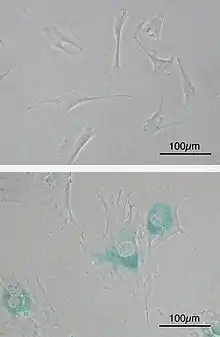
Bottom: MEFs became senescent after passages. Cells grow larger, flatten shape and expressed senescence-associated β-galactosidase (SABG, blue areas), a marker of cellular senescence.
The physiological importance for cell senescence has been attributed to prevention of carcinogenesis, and more recently, aging, development, and tissue repair.[6] Senescent cells contribute to the aging phenotype, including frailty syndrome, sarcopenia, and aging-associated diseases.[7] Senescent astrocytes and microglia contribute to neurodegeneration.[8]
Cellular mechanisms
Stress response and DNA damage
Mechanistically, replicative senescence can be triggered by a DNA damage response due to the shortening of telomeres. Cells can also be induced to senesce by DNA damage in response to elevated reactive oxygen species (ROS), activation of oncogenes, and cell-cell fusion. Normally, cell senescence is reached through a combination of a variety of factors (i.e., both telomere shortening and oxidative stress).[9] The DNA damage response (DDR) arrests cell cycle progression until DNA damage, such as double-strand breaks (DSBs), are repaired. Senescent cells display persistent DDR that appears to be resistant to endogenous DNA repair activities. The prolonged DDR activates both ATM and ATR DNA damage kinases. The phosphorylation cascade initiated by these two kinases causes the eventual arrest of the cell cycle. Depending on the severity of the DNA damage, the cells may no longer be able to undergo repair and either go through apoptosis or cell senescence.[5] Such senescent cells in mammalian culture and tissues retain DSBs and DDR markers.[10] It has been proposed that retained DSBs are major drivers of the aging process. Mutations in genes relating to genome maintenance has been linked with premature aging diseases, supporting the role of cell senescence in aging[11] (see DNA damage theory of aging).
Depletion of NAD+ can lead to DNA damage and cellular senescence in vascular smooth muscle cells.[12]
Although senescent cells can no longer replicate, they remain metabolically active and commonly adopt an immunogenic phenotype consisting of a pro-inflammatory secretome, the up-regulation of immune ligands, a pro-survival response, promiscuous gene expression (pGE), and stain positive for senescence-associated β-galactosidase activity.[13] Two proteins, senescence-associated beta-galactosidase and p16Ink4A, are regarded as biomarkers of cellular senescence. However, this results in a false positive for cells that naturally have these two proteins such as maturing tissue macrophages with senescence-associated beta-galactosidase and T-cells with p16Ink4A.[9]
Senescent cells can undergo conversion to an immunogenic phenotype that enables them to be eliminated by the immune system.[14] This phenotype consists of a pro-inflammatory secretome, the up-regulation of immune ligands, a pro-survival response, promiscuous gene expression (pGE) and stain positive for senescence-associated β-galactosidase activity.[13] The nucleus of senescent cells is characterized by senescence-associated heterochromatin foci (SAHF) and DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS).[15] Senescent cells affect tumour suppression, wound healing and possibly embryonic/placental development and a pathological role in age-related diseases.[16]
Role of telomeres
Telomeres are DNA tandem repeats at the end of chromosomes that shorten during each cycle of cell division.[17] Recently, the role of telomeres in cellular senescence has aroused general interest, especially with a view to the possible genetically adverse effects of cloning. The successive shortening of the chromosomal telomeres with each cell cycle is also believed to limit the number of divisions of the cell, contributing to aging. After sufficient shortening, proteins responsible for maintaining telomere structure, such as TRF2, are displaced, resulting in the telomere being recognized as a site of a double-strand break.[18] This induces replicative senescence.[19] Theoretically, it is possible upon the discovery of the exact mechanism of biological immortality to genetically engineer cells with the same capability. The length of the telomere strand has senescent effects; telomere shortening activates extensive alterations in alternative RNA splicing that produce senescent toxins such as progerin, which degrades tissue and makes it more prone to failure.[20]
Role of oncogenes
BRAFV600E and Ras are two oncogenes implicated in cellular senescence. BRAFV600E induces senescence through synthesis and secretion of IGFBP7.[21] Ras activates the MAPK cascade which results in increased p53 activation and p16INK4a upregulation.[22] The transition to a state of senescence due to oncogene mutations are irreversible and have been termed oncogene-induced senescence (OIS).[23]
Interestingly, even after oncogenic activation of a tissue, several researchers have identified a senescent phenotype. Researchers have identified a senescent phenotype in benign lesions of the skin carrying oncogenic mutations in neurofibroma patients with a defect that specifically causes an increase in Ras. This finding has been highly reproducible in benign prostate lesions, in melanocytic lesions of UV-irradiated HGF/SF-transgenic mice,[24] in lymphocytes and in the mammary gland from N-Ras transgenic mice,[25] and in hyperplasias of the pituitary gland of mice with deregulated E2F activity.[26] The key to these findings is that genetic manipulations that abrogated the senescence response led to full-blown malignancy in those carcinomas. As such, the evidence suggests senescent cells can be associated with pre-malignant stages of the tumor. Further, it has been speculated that a senescent phenotype might serve as a promising marker for staging. There are two types of senescence in vitro. The irreversible senescence which is mediated by INK4a/Rb and p53 pathways and the reversible senescent phenotype which is mediated by p53. This suggests that p53 pathway could be effectively harnessed as a therapeutic intervention to trigger senescence and ultimately mitigate tumorigenesis.[27]
p53 has been shown to have promising therapeutic relevance in an oncological context. In the 2007 Nature paper by Xue et al., RNAi was used to regulate endogenous p53 in a liver carcinoma model. Xue et al. utilized a chimaeric liver cancer mouse model and transduced this model with the ras oncogene. They took embryonic progenitor cells, transduced those cells with oncogenic ras, along with the tetracycline transactivator (tta) protein to control p53 expression using doxycycline, a tetracycline analog and tetracycline responsive short hairpin RNA (shRNA). In the absence of Dox, p53 was actively suppressed as the microRNA levels increased, so as Dox was administered, p53 microRNA was turned off to facilitate the expression of p53. The liver cancers that expressed Ras showed signs of senescence following p53 reactivation including an increase in senescence associated B-galactosidase protein. Even if the expression of p53 was transiently activated or deactivated, senescence via SA B-gal was observed. Xue et al. show that by briefly reactivating p53 in tumors without functional p53 activity, tumor regression is observed. The induction of cellular senescence was associated with an increase in inflammatory cytokines as is expected based on the SASP. The presence of both senescence and an increase in immune activity is able to regress and limit liver carcinoma growth in this mouse model.[28]
Signaling pathways
There are several reported signaling pathways that lead to cellular senescence including the p53 and p16Ink4a pathways.[23] Both of these pathways are activated in response to cellular stressors and lead to cell cycle inhibition. p53 activates p21 which deactivates cyclin-dependent kinase 2(Cdk 2). Without Cdk 2, retinoblastoma protein (pRB) remains in its active, hypophosphorylated form and binds to the transcription factor E2F1, an important cell cycle regulator.[29] This represses the transcriptional targets of E2F1, leading to cell cycle arrest after the G1 phase.
p16Ink4a also activates pRB, but through inactivation of cyclin-dependent kinase 4 (Cdk 4) and cyclin-dependent kinase 6 (Cdk 6). p16Ink4a is responsible for the induction of premature, stress-induced senescence.[29] This is not irreversible; silencing of p16Ink4a through promotor methylation or deletion of the p16Ink4a locus allows the cell to resume the cell cycle if senescence was initiated by p16Ink4a activation.
Senescence-associated secretory phenotype (SASP) gene expression is induced by a number of transcription factors, including C/EBPβ, of which the most important is NF-κB.[30] Aberrant oncogenes, DNA damage, and oxidative stress induce mitogen-activated protein kinases, which are the upstream regulators of NF-κB.[31]
Characteristics of senescent cells
Senescent cells are especially common in skin and adipose tissue.[7] Senescent cells are usually larger than non-senescent cells.[32] Transformation of a dividing cell into a non-dividing senescent cell is a slow process that can take up to six weeks.[32]
Senescent cells affect tumor suppression, wound healing and possibly embryonic/placental development, and play a pathological role in age-related diseases.[33] There are two primary tumor suppressor pathways known to mediate senescence: p14arf/p53 and INK4A/RB.[27] More specifically p16INK4a-pRb tumor suppressor and p53 are known effectors of senescence. Most cancer cells have a mutated p53 and p16INK4a-pRb, which allows the cancer cells to escape a senescent fate.[34] The p16 protein is a cyclin dependent kinase inhibitor (CDK) inhibitor and it activates Rb tumor suppressor.[35] p16 binds to CDK 4/6 to inhibit the kinase activity and inhibit Rb tumor suppressor via phosphorylation.[36] The Rb tumor suppressor has been shown to associate with E2F1 (a protein necessary for transcription) in its monophosphorylated form, which inhibits transcription of downstream target genes involved in the G1/S transition.[37] As part of a feedback loop, increased phosphorylation of Rb increases p16 expression that inhibits Cdk4/6. Reduced Cdk4/6 kinase activity results in higher levels of the hypo-phosphorylated (monophosphorylated) form of Rb, which subsequently leads to reduced levels of p16 expression.[36]
The removal of aggregated p16 INK 4A positive senescent cells can delay tissue dysfunction and ultimately extend life. In the 2011 Nature paper by Baker et al. a novel transgene, INK-ATTAC, was used to inducibly eliminate p16 INK4A-positive senescent cells by action of a small molecule-induced activation of caspase 8, resulting in apoptosis. A BubR1 H/H mouse model known to experience the clinicopathological characteristics of aging-infertility, abnormal curvature to the spine, sarcopenia, cataracts, fat loss, dermal thinning, arrhythmias, etc. was used to test the consequences of p16INK4a removal. In these mice p16 INK4a aggregates in aging tissues including the skeletal and eye muscle, and adipose tissues. Baker et al. found that if the senescent cells are removed, it is possible to delay age-associated disorders. Not only does p16 play an important role in aging, but also in auto-immune diseases like rheumatoid arthritis that progressively lead to mobility impairment in advanced disease.[35]
In the nervous system, senescence has been described in astrocytes and microglia, but is less understood in neurons.[38] Because senescence arrests cell division, studies of senescence in the brain were focused mainly on glial cells and less studies were focused on nondividing neurons.[39] Analyzing single nucleus RNA-Seq data from human brains suggested p19 as a marker for senescent neurons, which are strongly associated with neurons containing neurofibrillary tangle.[40]
SASP
The secretome of senescent cells is very complex. The products are mainly associated with inflammation, proliferation, and changes in the extracellular matrix.[41][34] A Senescence Associated Secretory Phenotype (SASP) consisting of inflammatory cytokines, growth factors, and proteases is another characteristic feature of senescent cells.[42] There are many SASP effector mechanisms that utilize autocrine or paracrine signalling. SASP induces an unfolded protein response in the endoplasmic reticulum because of an accumulation of unfolded proteins, resulting in proteotoxic impairment of cell function.[43] Autophagy is upregulated to promote survival.[43][44]
Considering cytokines, SASP molecules IL-6 and IL-8 are likely to cause senescence without affecting healthy neighbor cells. IL-1beta, unlike IL-6 or IL-8, is able to induce senescence in normal cells with paracrine signaling. IL-1beta is also dependent on cleavage of IL-1 by caspase-1, causing a pro-inflammatory response.[45] Growth factors, GM-CSF and VEGF also serve as SASP molecules.[46] From the cellular perspective, cooperation of transcriptional factors NF-κB and C/EBPβ increase the level of SASP expression.[34][47] Regulation of the SASP is managed through a transcription level autocrine feedback loop, but most importantly by a continuous DDR.[48][49] Proteins p53, p21, p16ink4a,[50] and Bmi-1 have been termed as major senescence signalling factors, allowing them to serve as markers.[51] Other markers register morphology changes, reorganization of chromatin, apoptosis resistance, altered metabolism, enlarged cytoplasm or abnormal shape of the nucleus.[52] SASPs have distinct effects depending on the cellular context, including inflammatory or anti-inflammatory and tumor or anti-tumor effects. While considered a pro-tumorogenic effect, they likely support already tumor-primed cells instead of shifting healthy cells into transformation.[52] Likewise, they operate as anti-tumor protectors[53] by facilitating the elimination of damaged cells by phagocytes. The SASP is associated with many age-related diseases, including type 2 diabetes and atherosclerosis.[9] This has motivated researchers to develop senolytic drugs to kill and eliminate senescent cells to improve health in the elderly.[9] The nucleus of senescent cells is characterized by senescence-associated heterochromatin foci (SAHF) and DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS).[52]
Clearance of senescent cells by the immune system
Due to the heterogeneous nature of senescent cells, different immune system cells eliminate different senescent cells.[54][55] Specific components of the senescence-associated secretory phenotype (SASP) factors secreted by senescent cells attract and activate different components of both the innate and adaptive immune system.[54]
Natural killer cells (NK cells) and macrophages play a major role in clearance of senescent cells.[56] Natural killer cells directly kill senescent cells, and produce cytokines which activate macrophages which remove senescent cells.[56] Senescent cells can be phagocytized by neutrophils as well as by macrophages.[57] Senolytic drugs which induce apoptosis in senescent cells rely on phagocytic immune system cells to remove the apoptosed cells.[55]
Natural killer cells can use NKG2D killer activation receptors to detect the MICA and ULBP2 ligands which become upregulated on senescent cells.[12][58] The senescent cells are killed using perforin pore-forming cytolytic protein.[57] CD8+ cytotoxic T-lymphocytes also use NKG2D receptors to detect senescent cells, and promote killing similar to NK cells.[57]
Aging of the immune system (immunosenescence) results in a diminished capacity of the immune system to remove senescent cells, thereby leading to an increase in senescent cells.[56] Chronic inflammation due to SASP from senescent cells can also reduce the capacity of the immune system to remove senescent cells.[57] T cells, B cells, and NK cells have all been reported to become senescent themselves.[59] Senescent-like aging CD8+ cytotoxic T-lymphocytes become more innate in structure and function, resembling NK cells.[60] Immune system cells can be recruited by SASP to senescent cells, after which the SASP from the senescent cells can induce the immune system cells to become senescent.[55]
Chimeric antigen receptor T cells have been proposed as an alternative means to senolytic drugs for the elimination of senescent cells.[55] Urokinase receptors have been found to be highly expressed on senescent cells, leading researchers to use chimeric antigen receptor T cells to eliminate senescent cells in mice.[61][62] Chimeric antigen receptor natural killer cells have been proposed as an allogeneic means of eliminating senescent cells.[63]
Transient senescence
It is important to recognize that cellular senescence is not inherently a negative phenomenon. During mammalian embryogenesis, programmed cellular senescence plays a role in tissue remodeling via macrophage infiltration and subsequent clearance of senescent cells.[64] A study on the mesonephros and endolymphatic sac in mice highlighted the importance of cellular senescence for eventual morphogenesis of the embryonic kidney and the inner ear, respectively.[64]
They serve to direct tissue repair and regeneration.[22] Cellular senescence limits fibrosis during wound closure by inducing cell cycle arrest in myofibroblasts once they have fulfilled their function.[22] When these cells have accomplished these tasks, the immune system clears them away. This phenomenon is termed acute senescence.[23] Senescence of hepatic stellate cells could prevent progression of liver fibrosis, although this has not been implemented as a therapy, and would carry the risk of hepatic dysfunction.[65]
The negative implications of cellular senescence present themselves in the transition from acute to chronic senescence. When the immune system cannot clear senescent cells at the rate at which senescent cells are being produced, possibly as a result of the decline in immune function with age, accumulation of these cells leads to a disruption in tissue homeostasis.[66]
Cellular senescence in mammalian disease
Transplantation of only a few (1 per 10,000) senescent cells into lean middle-aged mice was shown to be sufficient to induce frailty, early onset of aging-associated diseases, and premature death.[67]
Biomarkers of cellular senescence have been shown to accumulate in tissues of older individuals.[68] The accumulation of senescent cells in tissues of vertebrates with age is thought to contribute to the development of ageing-related diseases, including Alzheimer's disease, Amyotrophic lateral sclerosis, type 2 diabetes, and various cancers.[9][69][70][71]
Progeria is another example of a disease that may be related to cell senescence. The disease is thought to be caused by mutations in the DNA damage response, telomere shortening, or a combination of the two.[72] Progeroid syndromes are all examples of aging diseases where cell senescence appears to be implicated.
List of progeroid syndromes
- Hutchinson–Gilford progeria syndrome
- Rothmund–Thomson syndrome
- Werner syndrome
- Bloom syndrome
- Cockayne syndrome
- Xeroderma pigmentosum
- Trichothiodystrophy
- Xeroderma pigmentosum-Cockayne syndrome
- Restrictive dermopathy
- Mandibuloacral dysplasia
- Fanconi anaemia
- Seckel syndrome
- Ataxia telangiectasia
- Dyskeratosis congenita
- Hoyeraal-Hreidarsson syndrome
- Néstor-Guillermo progeria syndrome [73]
Senolytic drugs
Targeting senescent cells is a promising strategy to overcome age-related disease, simultaneous alleviate multiple comorbidities, and mitigate the effects of frailty. Removing the senescent cells by inducing apoptosis is the most straightforward option, and there are several agents that have been shown to accomplish this.[9] Some of these senolytic drugs take advantage of the senescent-cell anti-apoptotic pathways (SCAPs); knocking out expression of the proteins involved in these pathways can lead to the death of senescent cells, leaving healthy cells.[74]
Organisms lacking senescence
Cellular senescence is not observed in some organisms, including perennial plants, sponges, corals, and lobsters. In other organisms, where cellular senescence is observed, cells eventually become post-mitotic: they can no longer replicate themselves through the process of cellular mitosis (i.e., cells experience replicative senescence). How and why cells become post-mitotic in some species has been the subject of much research and speculation, but it has been suggested that cellular senescence evolved as a way to prevent the onset and spread of cancer.[75] Somatic cells that have divided many times will have accumulated DNA mutations and would be more susceptible to becoming cancerous if cell division continued. As such, it is becoming apparent that senescent cells undergo conversion to an immunologic phenotype that enables them to be eliminated by the immune system.[76]
See also
References
- Collado M, Blasco MA, Serrano M (July 2007). "Cellular senescence in cancer and aging". Cell. 130 (2): 223–33. doi:10.1016/j.cell.2007.07.003. PMID 17662938. S2CID 18689141.
- Hayat M (2014). Tumor dormancy, quiescence, and senescence, Volume 2: Aging, cancer, and noncancer pathologies. Springer. p. 188.
- Tollefsbol T (2010). Epigenetics of Aging. Springer. p. 227. ISBN 978-1-4419-0638-0.
- Shay JW, Wright WE (October 2000). "Hayflick, his limit, and cellular ageing". Nature Reviews. Molecular Cell Biology. 1 (1): 72–6. doi:10.1038/35036093. PMID 11413492. S2CID 6821048.
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (November 2010). "The essence of senescence". Genes & Development. 24 (22): 2463–79. doi:10.1101/gad.1971610. PMC 2975923. PMID 21078816.
- van Deursen JM (May 2014). "The role of senescent cells in ageing". Nature. 509 (7501): 439–46. Bibcode:2014Natur.509..439V. doi:10.1038/nature13193. PMC 4214092. PMID 24848057.
- Wyld L, Bellantuono I, Tchkonia T, Danson S, Kirkland JL (2020). "Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies". Cancers. 12 (8): e2134. doi:10.3390/cancers12082134. PMC 7464619. PMID 32752135.
- Rivera-Torres J, José ES (2019). "Src Tyrosine Kinase Inhibitors: New Perspectives on Their Immune, Antiviral, and Senotherapeutic Potential". Frontiers in Pharmacology. 10: 1011. doi:10.3389/fphar.2019.01011. PMC 6759511. PMID 31619990.
- Childs BG, Durik M, Baker DJ, van Deursen JM (December 2015). "Cellular senescence in aging and age-related disease: from mechanisms to therapy". Nature Medicine. 21 (12): 1424–35. doi:10.1038/nm.4000. PMC 4748967. PMID 26646499.
- Galbiati A, Beauséjour C, d'Adda di Fagagna F (April 2017). "A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues". Aging Cell. 16 (2): 422–427. doi:10.1111/acel.12573. PMC 5334542. PMID 28124509.
- White RR, Vijg J (September 2016). "Do DNA Double-Strand Breaks Drive Aging?". Molecular Cell. 63 (5): 729–38. doi:10.1016/j.molcel.2016.08.004. PMC 5012315. PMID 27588601.
- Song P, Zhao Q, Zou M (2020). "Targeting senescent cells to attenuate cardiovascular disease progression". Ageing Research Reviews. 60: 101072. doi:10.1016/j.arr.2020.101072. PMC 7263313. PMID 32298812.
- Campisi J (2013). "Aging, cellular senescence, and cancer". Annual Review of Physiology. 75: 685–705. doi:10.1146/annurev-physiol-030212-183653. PMC 4166529. PMID 23140366.
- Burton; Faragher (2015). "Cellular senescence: from growth arrest to immunogenic conversion". AGE. 37 (2): 27. doi:10.1007/s11357-015-9764-2. PMC 4365077. PMID 25787341.
- Rodier, F.; Campisi, J. (14 February 2011). "Four faces of cellular senescence". The Journal of Cell Biology. 192 (4): 547–56. doi:10.1083/jcb.201009094. PMC 3044123. PMID 21321098.
- Burton, Dominick G. A.; Krizhanovsky, Valery (31 July 2014). "Physiological and pathological consequences of cellular senescence". Cellular and Molecular Life Sciences. 71 (22): 4373–86. doi:10.1007/s00018-014-1691-3. PMC 4207941. PMID 25080110.
- Rivera T, Haggblom C, Cosconati S, Karlseder J (January 2017). "A balance between elongation and trimming regulates telomere stability in stem cells". Nature Structural & Molecular Biology. 24 (1): 30–39. doi:10.1038/nsmb.3335. PMC 5215970. PMID 27918544.
- Takai, Hiroyuki; Smogorzewska, Agata; de Lange, Titia (2003-09-02). "DNA Damage Foci at Dysfunctional Telomeres". Current Biology. 13 (17): 1549–1556. doi:10.1016/S0960-9822(03)00542-6. ISSN 0960-9822. PMID 12956959. S2CID 5626820.
- Victorelli, Stella; Passos, João F. (2017-07-01). "Telomeres and Cell Senescence - Size Matters Not". EBioMedicine. 21: 14–20. doi:10.1016/j.ebiom.2017.03.027. ISSN 2352-3964. PMC 5514392. PMID 28347656.
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, et al. (July 2011). "Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts". The Journal of Clinical Investigation. 121 (7): 2833–44. doi:10.1172/JCI43578. PMC 3223819. PMID 21670498.
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR (February 2008). "Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7". Cell. 132 (3): 363–74. doi:10.1016/j.cell.2007.12.032. PMC 2266096. PMID 18267069.
- Yun MH (2018-06-21). "Cellular senescence in tissue repair: every cloud has a silver lining". The International Journal of Developmental Biology. 62 (6–7–8): 591–604. doi:10.1387/ijdb.180081my. PMID 29938770.
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM (November 2014). "Senescence and apoptosis: dueling or complementary cell fates?". EMBO Reports. 15 (11): 1139–53. doi:10.15252/embr.201439245. PMC 4253488. PMID 25312810.
- Ha, L.; Ichikawa, T.; Anver, M.; Dickins, R.; Lowe, S.; Sharpless, N. E.; Krimpenfort, P.; DePinho, R. A.; Bennett, D. C.; Sviderskaya, E. V.; Merlino, G. (2007-06-18). "ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence". Proceedings of the National Academy of Sciences. 104 (26): 10968–10973. Bibcode:2007PNAS..10410968H. doi:10.1073/pnas.0611638104. ISSN 0027-8424. PMC 1904138. PMID 17576930.
- Braig, Melanie; Lee, Soyoung; Loddenkemper, Christoph; Rudolph, Cornelia; Peters, Antoine H. F. M.; Schlegelberger, Brigitte; Stein, Harald; Dörken, Bernd; Jenuwein, Thomas; Schmitt, Clemens A. (2005-08-04). "Oncogene-induced senescence as an initial barrier in lymphoma development". Nature. 436 (7051): 660–665. Bibcode:2005Natur.436..660B. doi:10.1038/nature03841. ISSN 1476-4687. PMID 16079837. S2CID 4373792.
- Lazzerini Denchi E, Attwooll C, Pasini D, Helin K (April 2005). "Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland". Molecular and Cellular Biology. 25 (7): 2660–72. doi:10.1128/MCB.25.7.2660-2672.2005. OCLC 842574443. PMC 1061636. PMID 15767672.
- Collado, Manuel; Blasco, Maria A.; Serrano, Manuel (2007-07-27). "Cellular senescence in cancer and aging". Cell. 130 (2): 223–233. doi:10.1016/j.cell.2007.07.003. ISSN 0092-8674. PMID 17662938. S2CID 18689141.
- Xue, Wen; Zender, Lars; Miething, Cornelius; Dickins, Ross A.; Hernando, Eva; Krizhanovsky, Valery; Cordon-Cardo, Carlos; Lowe, Scott W. (2007-02-08). "Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas". Nature. 445 (7128): 656–660. doi:10.1038/nature05529. ISSN 1476-4687. PMC 4601097. PMID 17251933.
- Ben-Porath I, Weinberg RA (May 2005). "The signals and pathways activating cellular senescence". The International Journal of Biochemistry & Cell Biology. 37 (5): 961–76. doi:10.1016/j.biocel.2004.10.013. PMID 15743671.
- Ghosh K, Capell BC (2016). "The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging". Journal of Investigative Dermatology. 136 (11): 2133–2139. doi:10.1016/j.jid.2016.06.621. PMC 5526201. PMID 27543988.
- Anerillas C, Abdelmohsen K, Gorospe M (2020). "Regulation of senescence traits by MAPKs". GeroScience. 42 (2): 397–408. doi:10.1007/s11357-020-00183-3. PMC 7205942. PMID 32300964.
- Kirkland JL, Tchkonia T (2020). "Senolytic Drugs: From Discovery to Translation". Journal of Internal Medicine. 288 (5): 518–536. doi:10.1111/joim.13141. PMC 7405395. PMID 32686219.
- Burton DG, Krizhanovsky V (November 2014). "Physiological and pathological consequences of cellular senescence". Cellular and Molecular Life Sciences. 71 (22): 4373–86. doi:10.1007/s00018-014-1691-3. PMC 4207941. PMID 25080110.
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. (June 2008). "Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network". Cell. 133 (6): 1019–31. doi:10.1016/j.cell.2008.03.039. PMID 18555778. S2CID 15295092.
- Baker, D.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.; Childs, B.; van de Sluis, B.; Kirkland, J.; van Deursen, J. (10 November 2011). "Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders". Nature. 479 (7372): 232–6. Bibcode:2011Natur.479..232B. doi:10.1038/nature10600. PMC 3468323. PMID 22048312.
- Rayess, Hani; Wang, Marilene B.; Srivatsan, Eri S. (2012-04-15). "Cellular senescence and tumor suppressor gene p16". International Journal of Cancer. 130 (8): 1715–1725. doi:10.1002/ijc.27316. ISSN 1097-0215. PMC 3288293. PMID 22025288.
- Narasimha, Anil M.; Kaulich, Manuel; Shapiro, Gary S.; Choi, Yoon J.; Sicinski, Piotr; Dowdy, Steven F. (2014-06-04). "Cyclin D activates the Rb tumor suppressor by mono-phosphorylation". eLife. 3. doi:10.7554/eLife.02872. ISSN 2050-084X. PMC 4076869. PMID 24876129.
- Vazquez-Villaseñor I, Garwood CJ, Heath PR, Simpson JE, Ince PG, Wharton SB (May 2019). "Expression of p16 and p21 in the frontal association cortex of ALS/MND brains suggests neuronal cell cycle dysregulation and astrocyte senescence in early stages of the disease". Neuropathology and Applied Neurobiology. 46 (2): 171–185. doi:10.1111/nan.12559. PMC 7217199. PMID 31077599.
- Chinta, Shankar J.; Woods, Georgia; Rane, Anand; Demaria, Marco; Campisi, Judith; Andersen, Julie K. (August 2015). "Cellular senescence and the aging brain". Experimental Gerontology. 68: 3–7. doi:10.1016/j.exger.2014.09.018. ISSN 1873-6815. PMC 4382436. PMID 25281806.
- Dehkordi, Shiva Kazempour; Walker, Jamie; Sah, Eric; Bennett, Emma; Atrian, Farzaneh; Frost, Bess; Woost, Benjamin; Bennett, Rachel E.; Orr, Timothy C.; Zhou, Yingyue; Andhey, Prabhakar S.; Colonna, Marco; Sudmant, Peter H.; Xu, Peng; Wang, Minghui; Zhang, Bin; Zare, Habil; Orr, Miranda E. (December 2021). "Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology". Nature Aging. 1 (12): 1107–1116. doi:10.1038/s43587-021-00142-3. ISSN 2662-8465. PMC 9075501. PMID 35531351.
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J (June 2008). "Chemokine signaling via the CXCR2 receptor reinforces senescence". Cell. 133 (6): 1006–18. doi:10.1016/j.cell.2008.03.038. PMID 18555777. S2CID 6708172.
- Malaquin N, Martinez A, Rodier F (September 2016). "Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype". Experimental Gerontology. 82: 39–49. doi:10.1016/j.exger.2016.05.010. PMID 27235851. S2CID 207584394.
- Soto-Gamez A, Quax WJ, Demaria M (2019). "Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions". Journal of Molecular Biology. 431 (15): 2629–2643. doi:10.1016/j.jmb.2019.05.036. PMID 31153901.
- Liu X, Ding J, Meng L (2018). "Oncogene-induced senescence: a double edged sword in cancer". Acta Pharmacologica Sinica. 39 (10): 1553–1558. doi:10.1038/aps.2017.198. PMC 6289471. PMID 29620049.
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (August 2013). "A complex secretory program orchestrated by the inflammasome controls paracrine senescence". Nature Cell Biology. 15 (8): 978–90. doi:10.1038/ncb2784. PMC 3732483. PMID 23770676.
- Information., Lawrence Berkeley National Laboratory. United States. Department of Energy. Office of Scientific and Technical (2008). Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. Lawrence Berkeley National Laboratory. OCLC 893411490.
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. (October 2011). "Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity". Genes & Development. 25 (20): 2125–36. doi:10.1101/gad.17276711. PMC 3205583. PMID 21979375.
- Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K, Gorospe M (August 2019). "Transcriptome signature of cellular senescence". Nucleic Acids Research. 47 (14): 7294–7305. doi:10.1093/nar/gkz555. PMC 6698740. PMID 31251810.
- Coppé JP, Desprez PY, Krtolica A, Campisi J (January 2010). "The senescence-associated secretory phenotype: the dark side of tumor suppression". Annual Review of Pathology. 5 (1): 99–118. doi:10.1146/annurev-pathol-121808-102144. PMC 4166495. PMID 20078217.
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (March 1997). "Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a". Cell. 88 (5): 593–602. doi:10.1016/s0092-8674(00)81902-9. PMID 9054499. S2CID 17518294.
- Schmitt CA (8 May 2018). "Senescence-associated reprogramming promotes cancer stemness" (PDF). Endocrine Abstracts. BioScientifica. 56 (7686): 96–100. Bibcode:2018Natur.553...96M. doi:10.1530/endoabs.56.s25.2. PMID 29258294.
- Salama R, Sadaie M, Hoare M, Narita M (January 2014). "Cellular senescence and its effector programs". Genes & Development. 28 (2): 99–114. doi:10.1101/gad.235184.113. PMC 3909793. PMID 24449267.
- Serrano M (November 2011). "Cancer: final act of senescence". Nature. 479 (7374): 481–2. Bibcode:2011Natur.479..481S. doi:10.1038/479481a. PMID 22113687. S2CID 36154048.
- Sagiv A, Krizhanovsky V (2013). "Immunosurveillance of senescent cells: the bright side of the senescence program". Biogerontology. 14 (6): 617–628. doi:10.1007/s10522-013-9473-0. PMID 24114507. S2CID 2775067.
- Song P, An J, Zou MH (2020). "Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases". Cells. 9 (3): E671. doi:10.3390/cells9030671. PMC 7140645. PMID 32164335.
- Antonangeli F, Zingoni A, Soriani A, Santoni A (2019). "Senescent cells: Living or dying is a matter of NK cells". Journal of Leukocyte Biology. 105 (6): 1275–1283. doi:10.1002/JLB.MR0718-299R. PMID 30811627. S2CID 73469394.
- Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL (2018). "Senescent cell clearance by the immune system: Emerging therapeutic opportunities". Seminars in Immunology. 40: 101275. doi:10.1016/j.smim.2019.04.003. PMC 7061456. PMID 31088710.
- Radosavljevic M, Cuillerier B, Wilson MJ, Clément O, Wicker S, Gilfillan S, Beck S, Trowsdale J, Bahram S (Jan 2002). "A cluster of ten novel MHC class I related genes on human chromosome 6q24.2-q25.3". Genomics. 79 (1): 114–23. doi:10.1006/geno.2001.6673. PMID 11827464.
- Frasca D (2018). "Senescent B cells in aging and age-related diseases: Their role in the regulation of antibody responses". Experimental Gerontology. 108: 55–58. doi:10.1016/j.exger.2017.07.002. PMC 5754260. PMID 28687479.
- Pereira BI, Akbar AN (2016). "Convergence of Innate and Adaptive Immunity during Human Aging". Frontiers in Immunology. 7: 445. doi:10.3389/fimmu.2016.00445. PMC 5095488. PMID 27867379.
- Wagner V, Gil J (2020). "T Cells Engineered to Target Senescence". Nature. 583 (7814): 37–38. Bibcode:2020Natur.583...37W. doi:10.1038/d41586-020-01759-x. PMID 32601490. S2CID 220260026.
- Amor C, Feucht J, Lowe SW (2020). "Senolytic CAR T cells reverse senescence-associated pathologies". Nature. 583 (7814): 127–132. Bibcode:2020Natur.583..127A. doi:10.1038/s41586-020-2403-9. PMC 7583560. PMID 32555459.
- Kale A, Sharma A, Stolzing A, Desprez P, Campisi J (2020). "Role of Immune Cells in the Removal of Deleterious Senescent Cells". Immunity & Ageing. 17: 16. doi:10.1186/s12979-020-00187-9. PMC 7271494. PMID 32518575.
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. (November 2013). "Programmed cell senescence during mammalian embryonic development". Cell. 155 (5): 1104–18. doi:10.1016/j.cell.2013.10.019. PMID 24238962.
- Zhang M, Serna-Salas S, Moshage H (2021). "Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives". Mechanisms of Ageing and Development. 199: 111572. doi:10.1016/j.mad.2021.111572. PMID 34536446. S2CID 237524296.
- Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T (October 2012). "Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans". Seminars in Immunology. 24 (5): 331–41. doi:10.1016/j.smim.2012.04.008. PMID 22560929.
- Khosla S, Farr JN, Tchkonia T, Kirkland JL (2020). "The role of cellular senescence in ageing and endocrine disease". Nature Reviews Endocrinology. 16 (5): 263–275. doi:10.1038/s41574-020-0335-y. PMC 7227781. PMID 32161396.
- Bernardes de Jesus B, Blasco MA (June 2012). "Assessing cell and organ senescence biomarkers". Circulation Research. 111 (1): 97–109. doi:10.1161/CIRCRESAHA.111.247866. PMC 4824275. PMID 22723221.
- Trias, Emiliano; Beilby, Pamela R.; Kovacs, Mariángeles; Ibarburu, Sofía; Varela, Valentina; Barreto-Núñez, Romina; Bradford, Samuel C.; Beckman, Joseph S.; Barbeito, Luis (2019). "Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS". Frontiers in Aging Neuroscience. 11: 42. doi:10.3389/fnagi.2019.00042. ISSN 1663-4365. PMC 6403180. PMID 30873018.
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. (May 2019). "Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model". Nature Neuroscience. 22 (5): 719–728. doi:10.1038/s41593-019-0372-9. PMC 6605052. PMID 30936558.
- Aguayo-Mazzucato C, Andle J, Lee TB, Midha A, Talemal L, Chipashvili V, et al. (July 2019). "Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes". Cell Metabolism. 30 (1): 129–142.e4. doi:10.1016/j.cmet.2019.05.006. PMC 6610720. PMID 31155496.
- Sinha JK, Ghosh S, Raghunath M (May 2014). "Progeria: a rare genetic premature ageing disorder". The Indian Journal of Medical Research. 139 (5): 667–74. PMC 4140030. PMID 25027075.
- Carrero D, Soria-Valles C, López-Otín C (July 2016). "Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells". Disease Models & Mechanisms. 9 (7): 719–35. doi:10.1242/dmm.024711. PMC 4958309. PMID 27482812.
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD (October 2017). "The Clinical Potential of Senolytic Drugs". Journal of the American Geriatrics Society. 65 (10): 2297–2301. doi:10.1111/jgs.14969. PMC 5641223. PMID 28869295.
- Sahu S, Dattani A, Aboobaker AA (October 2017). "Secrets from immortal worms: What can we learn about biological ageing from the planarian model system?". Seminars in Cell & Developmental Biology. Science communication in the field of fundamental biomedical research. 70: 108–121. doi:10.1016/j.semcdb.2017.08.028. PMID 28818620.
- Burton DG, Faragher RG (2015). "Cellular senescence: from growth arrest to immunogenic conversion". Age. 37 (2): 27. doi:10.1007/s11357-015-9764-2. PMC 4365077. PMID 25787341.
Further reading
- Hayflick L, Moorhead PS (December 1961). "The serial cultivation of human diploid cell strains". Experimental Cell Research. 25 (3): 585–621. doi:10.1016/0014-4827(61)90192-6. PMID 13905658.
- Hayflick L (March 1965). "The limited in vitro lifetime of human diploid cell strains". Experimental Cell Research. 37 (3): 614–36. doi:10.1016/0014-4827(65)90211-9. PMID 14315085.
External links
 Media related to Cellular senescence at Wikimedia Commons
Media related to Cellular senescence at Wikimedia Commons