Sic1
Sic1, a protein, is a stoichiometric inhibitor[1] of Cdk1-Clb (B-type cyclins) complexes in the budding yeast Saccharomyces cerevisiae. Because B-type cyclin-Cdk1 complexes are the drivers of S-phase initiation, Sic1 prevents premature S-phase entry.[2] Multisite phosphorylation of Sic1 is thought to time Sic1 ubiquitination and destruction, and by extension, the timing of S-phase entry.[3]
| Sic1 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | Sic1 | ||||||
| Alt. symbols | YLR079W, SDB25, SIC1_YEAST, CDK inhibitor p40 | ||||||
| NCBI gene | 850768 | ||||||
| UniProt | P38634 | ||||||
| |||||||
Cell cycle control
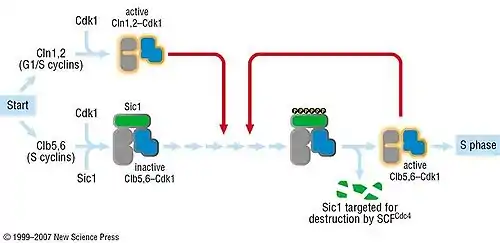
In the G1 phase of the cell cycle, Sic1 binds tightly to the Cdc28-Clb complex and inhibits it.[4] Low Cdc28-Clb activity leads to the disassembly of the mitotic spindle, the assembly of the prereplicative complex and initiation of bud formation in yeast.
At the START point in the yeast cell cycle, the G1-cyclins Cln3, Cln1 and Cln 2 activate Cdc28. The activated complex will phosphorylate Sic1 at multiple sites which leads to its degradation by the SCF complex.[5] When Sic1 is degraded, the Cdc28-Clb complex is no longer inhibited and the cell can enter the S/M-phase. Thus Sic1 inactivation is essential for transition into S phase (Fig.1).
Cdc28 in complex with B-type cyclin (Cdc28-Clb) phosphorylates Swi5, the transcription factor of Sic1. This promotes the export of Swi5 from the nucleus to the cytoplasm and avoids further transcription of the cdk inhibitor. Cdc28-Clb also phosphorylates any Sic1 molecules still available and triggers their ubiquitin-dependent degradation, exactly like Cdc28-Cln.[4] High Cdc28-Clb levels also initiate DNA replication and duplication of the spindle pole bodies (SPBs). Then the metaphase spindle assembles and chromosome segregation can occur. The transcription of Sic1 starts during telophase, mediated by Swi5. Aca2 is another transcription factor of Sic1, but remains inactive until G1.[6] At the end of mitosis, Sic1 is involved in the inactivation of Cdc28-Clb.[7]
Ubiquitin-dependent degradation
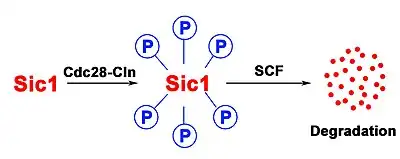
In order to be recognized by Cdc4 of the SCF complex, Sic1 has to be phosphorylated, often by Cyclin-Cdk complexes, at least at 6 of the 9 cdk sites (Fig. 2).[8] Sic1 can also be phosphorylated by other kinases, such as Pho85-Pc11, a kinase which becomes essential when Cln1 and Cln2 are absent.[9] Sic1 has also a role in the response to osmostress. The stress-activated protein kinase (SAPK) Hog1 phosphorylates Sic1 at a single residue at the carboxyl terminus. This leads to downregulation of cyclin expression and Sic1 stabilization which arrests the cell cycle.[10]
Phosphorylation
Sic1 needs to be phosphorylated at multiple sites for ubiquitination-driven degradation (Fig. 2). The multiple phosphorylations are required for Sic1 to be recruited by Cdc4 to the SCF complex.[11] The Cdc4 substrate recognition mechanism includes the interaction with consensus binding motifs on the surface of the folded and phosphorylated Sic1, the so-called Cdc4 phospho-degrons (CPD). It has been shown that the optimal consensus sequence for Cdc4 is a phosphorylated serine or threonine followed by a proline and a basic amino acid. However, none of the CPDs on the surface of the Sic1 show such a composition. Therefore, multiple phosphorylation of Sic1 is necessary to get high-affinity binding to Cdc4.[8] Although this mechanism looks inefficient, it provides advantages for a cell because it is possible to measure the environmental Cln/cdc28 concentration. The number of phosphorylated sites corresponds to the concentration of Cln/cdc28 and Sic1 could be considered as a sensor for this protein. In contrast to the many sharp transitions of ultrasensitive kinase cascade feedback loops, this mechanism allows fine tuned regulation.[8] Moreover, because multiple phosphorylations are required, the probability that Sic1 is degraded by random is small. Using multiple phosphorylation of Sic1, the cell evolved a strategy to highly regulate the onset of DNA replication that is absolutely vital to provide genetic stability.
A simplified understanding of the regulation of Sic1 degradation involves the phosphorylation of multiple CDK sites, which consist of optimal and suboptimal consensus phosphorylation motifs. Recent studies conducted by Koivomagi et al. have revealed the many intricacies of the multi-phosphorylation reaction between the cyclin-CDK complex and the Sic1 protein. These studies unveil the important characteristics of the Sic1 CDK phosphorylation sites, which include priming sites, binding sites, degron pairs, distancing of phosphorylation sites, and relative site location. In addition, the studies also emphasize the influence of other factors on Sic1 phosphorylation, including the Cks1 phospho-binding pocket, cyclin docking motifs, and Cdk1 active site specificity. All of these mechanisms contribute to the dynamics of the sequence of events leading to Sic1 degradation and initiation of S-phase.[12][13]
Function
Apart from being an often-overlooked component of the Cdk1 cyclin complex, Cks1 is critical for Sic1 multi-phosphorylation and degradation. The phospho-binding pocket of Cks1 is capable of binding independently to phosphorylated CDK sites on Sic1. Additionally, the binding affinity of Cks1 for phosphoserines is extremely weak, essentially making Cks1 binding dependent on the presence of phosphothreonines only. Thus, in Sic1 mutants with one Cdk1 phosphorylation site or only phosphoserines present, Cks1 is unable to properly bind to the substrate and promote Sic1 multi-phosphorylation. This provides a strong argument for a processive phosphorylation mechanism instead of the previous theory of a random distributive phosphorylation model.[8] In addition to requiring threonine, Cks1 binding to Sic1 can be enhanced with the introduction of a proline residue at the -2 position relative to the threonine residue.[12][13]
Site positioning
Sic1 is a molecule with disordered regions, which aids in the manipulation of phosphorylation site distances. For the following findings, Koivomagi et al. utilized a Sic1 construct with a T33 optimal consensus motif, acting as the primary phosphorylation site, and a suboptimal motif, acting as a secondary site.[12][13]
When limiting observations to only double-phosphorylated Sic1 constructs, a two-step phosphorylation process was observed, where the first step was primary site phosphorylation. However, the secondary site must be located towards the C-terminus of the protein relative to the primary site for phosphorylation to occur. Secondary site phosphorylation is also sensitive to positioning. Peak phosphorylation rates were found between the +12 to +16 amino acid distances, with a distinct increase around the +10 to +12 range and gradual decrease across the +20 to +30 range. The introduction of the -2 proline residue enhances phosphorylation both in vitro by expanding the peak phosphorylation range, but does not increase phosphorylation activity at distances less than +10. This expansion of the peak phosphorylation range could possibly be attributed to enhanced binding of the priming site to Cks1.[12][13]
A simple Sic1 construct containing 5 phosphorylation residues (1 priming site and two phosphodegron pairs) revealed that any slight movement of the priming site can have significant effects on cell cycle progression. The priming site should be within the +12 to +16 range of both residues in the phosphodegron pair to maximize phosphorylation.[12][13]
Directionality
Sic1 phosphorylation is initiated by the G1 cyclins, Cln1,2, and then completed by S-phase cyclins, Clb5,6 (Fig. 1). The docking motifs of the cyclin participate in Sic1 phosphorylation dynamics. S-phase cyclins use RXL docking while G1 cyclins use LLPP docking. Sic1 phosphorylation increases when the RXL motif of Clb5 is +16 to +20 positions relative to the optimal CDK motif. RXL positioning located N-terminal to the motif led to negligible amounts of phosphorylation. In contrast, moving the LLPP motif away from the priming site increases Cln2 phosphorylation, regardless of directionality.[12][13]
Processivity
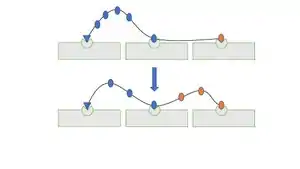
Cks1-dependent multi-phosphorylation occurs in a processive or semi-processive manner, evidenced by the lack of intermediate Sic1 phosphorylation states in normal cells. This processivity is also dependent on the presence of the cyclin docking site since increasing the numbers of mutations in this site decreases the net phosphorylation rate. Processive phosphorylation has two plausible mechanisms where a single binding event leads to the phosphorylation of two or more sites. The first mechanism proposes that, without dissociating from the enzyme complex, the primary site is phosphorylated and immediately shifted from the active site to the Cks1 binding pocket to allow for the additional phosphorylation of other CDK sites. The second mechanism proposes that the phosphorylated primary site binds to another location and is continuously bound while other CDK sites bind to the active site in a sequential manner for multi-phosphorylation. Simulations predict that the probability of a second phosphorylation event after the first, without dissociation, is 40% and 20-40% for the first and second mechanism, respectively.[12][13]
Sic1 is targeted for degradation by SCF (Cdc4), which recognizes Sic1 phosphodegron pairs. These phosphodegron pairs are closely positioned paired phosphorylation residues that each have strong affinities for Cdc4. In a Sic1 construct with the S69/S76/S80 cluster, processive phosphorylation of these phosphodegron pairs are reliant on Cdk1 sites. Clb5 processivity is dependent on the T5 and T33 sites, while Cln2 processivity is dependent on T5. Reintroduction of various residues led to the discovery of the T33 residue serving as a docking site for the T45/T48 phosphodegron pair, which is able to promote Sic1 degradation to a certain extent in the absence of other phosphodegron pairs.[12][13]
Mechanism
The following is a proposed mechanism by Koivomagi et al. of the in vivo cascade to promote Sic1 phosphorylation and degradation.
In late G1, Sic1 is inhibiting the Clb5-Cdk1 complex, simultaneously inhibiting its own degradation. The phosphorylation cascade proceeds by Cln2-Cdk1 phosphorylation of the T5 priming site. Following this, the T33, T45, and S76 residues are phosphorylated by Cln2-Cdk1, but no degron pairs are phosphorylated. However, these phosphorylated sites enhance Clb5-Cdk1 docking, leading to increased Sic1 phosphorylation at suboptimal sites and a positive feedback loop where Clb5-Cdk1 inhibition is continually decreased while Sic1 degradation is increased.[12][13]
Sic1 homologue in human and diseases
The protein p27Kip1 is a human homologue of Sic1, both having a conserved inhibitory domain,[14] but p27Kip1 inhibits G1 cyclins and not cyclin B.
There are several human diseases that are linked to p27Kip1 and other cyclin kinase inhibitors:
- All Papillary microcarcinomas (PMCs) of the thyroid have a lower expression of p27Kip1 than normal thyroid tissue. Additionally, the expression of p27Kip1 in more aggressive, metastasising Papillary microcarcinomas is strongly reduced compared to nonmetastasing microcarcinomas. These results suggest that p27Kip1 acts as a tumor suppressor.[15]
- Kaposi's sarcoma is a type of cancer which appears in combination with AIDS and presumably is caused by the human herpesvirus 8 (HHV8). This virus expresses a viral cyclin which builds a complex with Cdk6. This KSHV-cyclin-Cdk6 complex phosphorylates and destabilises p27Kip1, which results in a low level of p27Kip1. This suggests that the degradation of p27Kip1 is associated with the development of the tumors.[16]
- Patients with a gastric adenocarcinoma (stomach cancer) have a higher chance of survival if the tumor has high p27Kip1 expression. Low p27Kip1 expression can lead to tumor de-differentiation, increased penetration through the gastric wall, lymph node metastasis and advanced tumor stage.[17]
Thus, the human Cdk inhibitor p27Kip1 is a potential tumor suppressor protein. If its expression is reduced, the result might be unregulated progression from G1 to S-phase which deregulates cell division and simplifies the formation of tumors.
References
- Schwob E, Böhm T, Mendenhall MD, Nasmyth K (October 1994). "The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae". Cell. 79 (2): 233–44. doi:10.1016/0092-8674(94)90193-7. PMID 7954792. S2CID 34939988.
- Morgan DO (1997). The Cell Cycle: Principles of Control. London: New Science Press. pp. 200–1. ISBN 978-0-87893-508-6.
- Tripodi F, Zinzalla V, Vanoni M, Alberghina L, Coccetti P (August 2007). "In CK2 inactivated cells the cyclin dependent kinase inhibitor Sic1 is involved in cell-cycle arrest before the onset of S phase". Biochemical and Biophysical Research Communications. 359 (4): 921–7. doi:10.1016/j.bbrc.2007.05.195. PMID 17574209.
- Cross FR, Schroeder L, Bean JM (July 2007). "Phosphorylation of the Sic1 inhibitor of B-type cyclins in Saccharomyces cerevisiae is not essential but contributes to cell cycle robustness". Genetics. 176 (3): 1541–55. doi:10.1534/genetics.107.073494. PMC 1931548. PMID 17483408.
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ (October 1997). "Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase". Science. 278 (5337): 455–60. Bibcode:1997Sci...278..455V. doi:10.1126/science.278.5337.455. PMID 9334303.
- Toyn JH, Johnson AL, Donovan JD, Toone WM, Johnston LH (January 1997). "The Swi5 transcription factor of Saccharomyces cerevisiae has a role in exit from mitosis through induction of the cdk-inhibitor Sic1 in telophase". Genetics. 145 (1): 85–96. doi:10.1093/genetics/145.1.85. PMC 1207787. PMID 9017392.
- Calzada A, Sacristán M, Sánchez E, Bueno A (July 2001). "Cdc6 cooperates with Sic1 and Hct1 to inactivate mitotic cyclin-dependent kinases". Nature. 412 (6844): 355–8. Bibcode:2001Natur.412..355C. doi:10.1038/35085610. PMID 11460169. S2CID 4410112.
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M (November 2001). "Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication". Nature. 414 (6863): 514–21. Bibcode:2001Natur.414..514N. doi:10.1038/35107009. PMID 11734846. S2CID 16924667.
- Nishizawa M, Kawasumi M, Fujino M, Toh-e A (September 1998). "Phosphorylation of sic1, a cyclin-dependent kinase (Cdk) inhibitor, by Cdk including Pho85 kinase is required for its prompt degradation". Molecular Biology of the Cell. 9 (9): 2393–405. doi:10.1091/mbc.9.9.2393. PMC 25506. PMID 9725902.
- Escoté X, Zapater M, Clotet J, Posas F (October 2004). "Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1". Nature Cell Biology. 6 (10): 997–1002. doi:10.1038/ncb1174. PMID 15448699. S2CID 19846318.
- Coccetti P, Zinzalla V, Tedeschi G, Russo GL, Fantinato S, Marin O, Pinna LA, Vanoni M, Alberghina L (August 2006). "Sic1 is phosphorylated by CK2 on Ser201 in budding yeast cells". Biochemical and Biophysical Research Communications. 346 (3): 786–93. doi:10.1016/j.bbrc.2006.05.171. PMID 16777072.
- Kõivomägi M, Ord M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ER, Rubin SM, Loog M (December 2013). "Multisite phosphorylation networks as signal processors for Cdk1". Nature Structural & Molecular Biology. 20 (12): 1415–24. doi:10.1038/nsmb.2706. PMC 3855452. PMID 24186061.
- Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, Loog M (October 2011). "Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase". Nature. 480 (7375): 128–31. Bibcode:2011Natur.480..128K. doi:10.1038/nature10560. PMC 3228899. PMID 21993622.
- Barberis M, De Gioia L, Ruzzene M, Sarno S, Coccetti P, Fantucci P, Vanoni M, Alberghina L (May 2005). "The yeast cyclin-dependent kinase inhibitor Sic1 and mammalian p27Kip1 are functional homologues with a structurally conserved inhibitory domain". The Biochemical Journal. 387 (Pt 3): 639–47. doi:10.1042/BJ20041299. PMC 1134993. PMID 15649124.
- Khoo ML, Freeman JL, Witterick IJ, Irish JC, Rotstein LE, Gullane PJ, Asa SL (March 2002). "Underexpression of p27/Kip in thyroid papillary microcarcinomas with gross metastatic disease". Archives of Otolaryngology–Head & Neck Surgery. 128 (3): 253–7. doi:10.1001/archotol.128.3.253. PMID 11886339.
- Mann DJ, Child ES, Swanton C, Laman H, Jones N (February 1999). "Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus". The EMBO Journal. 18 (3): 654–63. doi:10.1093/emboj/18.3.654. PMC 1171158. PMID 9927425.
- Nitti D, Belluco C, Mammano E, Marchet A, Ambrosi A, Mencarelli R, Segato P, Lise M (December 2002). "Low level of p27(Kip1) protein expression in gastric adenocarcinoma is associated with disease progression and poor outcome". Journal of Surgical Oncology. 81 (4): 167–75, discussion 175–6. doi:10.1002/jso.10172. PMID 12451619. S2CID 5877623.