Single-cell DNA template strand sequencing
Single-cell DNA template strand sequencing, or Strand-seq, is a technique for the selective sequencing of a daughter cell's parental template strands.[1] This technique offers a wide variety of applications, including the identification of sister chromatid exchanges in the parental cell prior to segregation, the assessment of non-random segregation of sister chromatids, the identification of misoriented contigs in genome assemblies, de novo genome assembly of both haplotypes in diploid organisms including humans, whole-chromosome haplotyping, and the identification of germline and somatic genomic structural variation, the latter of which can be detected robustly even in single cells.
Background
Strand-seq (single-cell and single-strand sequencing) was one of the first single-cell sequencing protocols described in 2012.[1] This genomic technique selectively sequencings the parental template strands in single daughter cells DNA libraries.[1] As a proof of concept study, the authors demonstrated the ability to acquire sequence information from the Watson and/or Crick chromosomal strands in an individual DNA library, depending on the mode of chromatid segregation; a typical DNA library will always contain DNA from both strands. The authors were specifically interested in showing the utility of strand-seq in detecting sister chromatid exchanges (SCEs) at high-resolution. They successfully identified eight putative SCEs in the murine (mouse) embryonic stem (meS) cell line with resolution up to 23 bp. This methodology has also been shown to hold great utility in discerning patterns of non-random chromatid segregation, especially in stem cell lineages. Furthermore, SCEs have been implicated as diagnostic indicators of genome stress, information that has utility in cancer biology. Most research on this topic involves observing the assortment of chromosomal template strands through many cell development cycles and correlating non-random assortment with particular cell fates. Single-cell sequencing protocols were foundational in the development of this technique, but they differ in several aspects.
Methodology
Similar methods
Past methods have been used to track the inheritance patterns of chromatids on a per-strand basis and elucidate the process of non-random segregation:
Pulse-chase
Pulse-chase experiments have been used for determining the segregation patterns of chromosomes in addition to studying other time-dependent cellular processes.[2] Briefly, pulse-chase assays allow researchers to track radioactively labelled molecules in the cell. In experiments used to study non-random chromosome assortment, stem cells are labeled or "pulsed" with a nucleotide analog that is incorporated in the replicated DNA strands.[3] This allows the nascent stands to be tracked through many rounds of replication. Unfortunately, this method is found to have poor resolution as it can only be observed at the chromatid level.
Chromosome-orientation fluorescence in situ hybridization (CO-FISH)
CO-FISH, or strand-specific fluorescence in situ hybridization, facilitates strand-specific targeting of DNA with fluorescently-tagged probes.[4] It exploits the uniform orientation of major satellites relative to the direction of telomeres, thus allowing strands to be unambiguously designated as "Watson" or "Crick" strands. Using unidirectional probes that recognize major satellite regions, coupled to fluorescently labelled dyes, individual strands can be bound.[4] To ensure that only the template strand is labelled, the newly formed strands must be degraded by BrdU incorporation and photolysis. This protocol offers improved cytogenetic resolution, allowing researchers to observe single strands as opposed to whole chromatids with pulse-chase experiments. Moreover, non-random segregation of chromatids can be directly assayed by targeting major satellite markers.
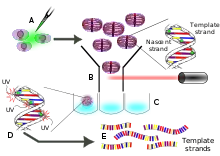
Wet lab protocols
Cells of interest are cultured either in vivo or in vitro. During S-phase cells are treated with bromodeoxyuridine (BrdU) which is then incorporated into their nascent DNA, acting as a substitute for thymidine. After at least one replication event has occurred, the daughter cells are synchronized at the G2 phase and individually separated by fluorescence-activated cell sorting (FACS). The cells are directly sorted into lysis buffer and their DNA is extracted. Having been arrested at a specified number of generations (usually one), the inheritance patterns of sister chromatids can be assessed. The following methods concentrate on the DNA sequencing of a single daughter cell's DNA. At this point the chromosomes are composed of nascent strands with BrdU in place of thymidine and the original template strands are primed for DNA sequencing library preparation. Since this protocol was published in 2012,[1] the canonical methodology is only well described for Illumina sequencing platforms; the protocol could very easily be adapted for other sequencing platforms, depending on the application. Next, the DNA is incubated with a special dye such that when the BrdU-dye complex is excited by UV light, nascent strands are nicked by photolysis. This process inhibits polymerase chain reaction (PCR) amplification of the nascent strand, allowing only the parental template strands to be amplified. Library construction proceeds as normal for Illumina paired-end sequencing. Multiplexing PCR primers are then ligated to the PCR amplicons with hexamer barcodes identifying which cell each fragment they are derived from. Unlike single cell sequencing protocols, Strand-seq does not utilize multiple displacement amplification or MALBAC for DNA amplification. Rather, it is solely dependent on PCR.
Bioinformatic processing
_Output.png.webp)
The majority of current applications for Strand-seq start by aligning sequenced reads to a reference genome. Alignment can be performed using a variety of short-read aligners such as BWA and Bowtie.[5][6] By aligning Strand-seq reads from a single cell to the reference genome, the inherited template strands can be determined. If the cell was sequenced after more than one generation, a pattern of chromatid assortment can be ascertained for the particular cell lineage at hand. The Bioinformatic Analysis of Inherited Templates (BAIT) was the first bioinformatic software to exclusively analyze reads generated from the Strand-seq methodology.[7] It begins by aligning the reads to a reference sequence, binning the genome into sections, and finally counting the number of Watson and Crick reads falling within each bin. From here, BAIT enables the identification of SCE events, misoriented contigs in the reference genome, aneuploid chromosomes and modes of sister chromatid segregation. It can also aid in assembling early-build genomes and assigning orphan scaffolds to locations within late-build genomes. Following BAIT, numerous bioinformatics tools have recently been introduced that use Strand-seq data for a variety of applications (see, for example, the following sections on haplotyping, de novo genome assembly, and discovery of structural variations in single cells, with reference to the respective linked articles).
Limitations
Strand-seq requires cells undergoing cell division for BrdU labeling, and thus is not applicable to formalin-fixed specimens or non-dividing cells. But it may be applied to normal mitotic cells and tissues, organoids, as well as leukemia and tumor samples using fresh or frozen primary specimens. Strand-seq is using Illumina sequencing, and applications that require sequence information from different sequencing technologies require new protocols, or alternatively integration of data generated using distinct sequencing platforms as recently show-cased.[8][9]
Authors from the initial papers describing Strand-seq showed that they were able to attain a 23bp resolution for mapping SCEs, and other large chromosomal abnormalities are likely to share this mapping resolution (if breakpoint fine-mapping is performed). Resolution, however, is dependent on a combination of the sequencing platform used, library preparation protocols, and the number of cells analysed as well as the depth of sequencing per cell. However, it would be sensical for precision to further increase with sequencing technologies that don't incur errors in homopolymeric repeats.
Applications and utility
Identifying sister chromatid exchanges
Strand-seq was initially proposed as a tool to identify sister chromatid exchanges.[1] Being a process that is localized to individual cells, DNA sequencing of more than one cell would naturally scatter these effects and suggest an absence of SCE events. Moreover, classic single cell sequencing techniques are unable to show these events due to heterogeneous amplification biases and dual-strand sequence information, thereby necessitating Strand-seq. Using the reference alignment information, researchers can identify an SCE if the directionality of an inherited template strand changes.
Identifying misoriented contigs
Misoriented contigs are present in reference genomes at significant rates (ex. 1% in the mouse reference genome).[1][10] Strand-seq, in contrast to conventional sequencing methods, can detect these misorientations.[1] Misoriented contigs are present where strand inheritance changes from one homozygous state to the other (ex. WW to CC, or CC to WW). Moreover, this state change is visible in every Strand-seq library, reinforcing the presence of a misoriented contig.[7]
Identifying non-random segregation of sister chromatids
Prior to the 1960s, it was assumed that sister chromatids were segregated randomly into daughter cells. However, non-random segregation of sister chromatids has been observed in mammalian cells ever since.[11][12] There have been a few hypotheses proposed to explain the non-random segregation, including the Immortal Strand Hypothesis and the Silent Sister Hypothesis, one of which may hopefully be verified by methods involving Strand-seq.
‘’Immortal Strand Hypothesis’’
Mutations occur every time a cell divides. Certain long-lived cells (ex. stem cells) may be particularly affected by these mutations. The Immortal Strand Hypothesis proposes that these cells avoid mutation accumulation by consistently retaining parental template strands[9]. For this hypothesis to be true, sister chromatids from each and every chromosome must segregate in a non-random fashion. Additionally, one cell will retain the exact same set of template strands after each division, giving the rest to the other cell products of the division.[13]
‘’Silent Sister Hypothesis’’
This hypothesis states that sister chromatids have differing epigenetic signatures, thereby also differing expression regulation. When replication occurs, non-random segregation of sister chromatids ensures the fates of the daughter cells.[14] Assessing the validity of this hypothesis would require a joint analysis of Strand-seq and gene expression profiles for both daughter cells.[13]
Discovery of structural variations & aneuploid chromosomes
The output of BAIT shows the inheritance of parental template strands along the genome.[7] Normally, two template strands are inherited for each autosome, and any deviation from this number indicates an instance of aneuploidy, which can be visualised in single cells.
Inversions are a class of copy-number balanced structural variation, which lead to a change in strand directionality readily visualised by Strand-seq. Strand-seq can hence be used to readily detect polymorphic inversions in humans and primates, including Megbase-sized events embedded in large segmental duplications known to be inaccessible to Illumina sequencing.[15][16]
A study published online in 2019 further demonstrated that using Strand-seq, all classes of structural variation ≥200kb including deletions, duplications, inversions, inverted duplications, balanced translocations, unbalanced translocations, breakage-fusion-bridge cycle mediated complex DNA rearrangements, and chromothripsis events are sensitively detected in single cells or subclones, using single-cell tri-channel processing (scTRIP). scTRIP works via joint modelling of read-orientation, read-depth, and haplotype-phase to discover SVs in single cells.[17] Using scTRIP, structural variants are resolved by chromosome-length haplotype which confers higher sensitivity and specificity for single-cell structural variant calling than other current technologies.[17] Since scTRIP does not require reads (or read pairs) transversing the boundaries (or breakpoints) of structural variants in single cells for variant calling, it does not suffer from known artefacts of single-cell methods based on whole genome amplification (i.e. so-called read chimera) which tend to confound structural variation analysis in single cells.[17]
Haplotyping, genome assembly & generation of high-resolution human genetic variation maps
Early-build genomes are quite fragmented, with unordered and unoriented contigs. Using Strand-seq provides directionality information to accompany the sequence, which ultimately helps resolve the placement of contigs. Contigs present in the same chromosome will exhibit the same directionality, provided SCE events have not occurred. Conversely, contigs present in different chromosomes will only exhibit the same directionality in 50% of the Strand-seq libraries.[7] Scaffolds, successive contigs intersected by a gap, can be localized in the same manner.[7]
The same principle of using strand direction to distinguish large DNA molecules enables the use of Strand-seq as a tool to construct whole-chromosome haplotypes of genetic variation, from telomere to telomere.[18]
Recent reports have shown that Strand-seq can be computationally integrated with long-read sequencing technology, with the unique advantages of both technologies enabling the generation of highly contiguous haplotype-resolved de novo human genome assemblies.[8] These genomic assemblies integrate all forms of genetic variation including single nucleotide variants, indels and structural variation even across complex genomic loci, and have recently been applied to generate comprehensive haplotype-aware maps of structural variation in a diversity panel of humans from distinct ancestries.[9]
Considerations
The possibility that BrdU being substituted for thymine in the genomic DNA could induce double stranded chromosomal breaks and specifically resulting in SCEs has been previously discussed in the literature.[19][20] Additionally, BrdU incorporation has been suggested to interfere with strand segregation patterns.[13] If this is the case, there would be an inflation in false positive SCEs which may be annotated. Therefore, many cells should be analyzed using the Strand-seq protocol to ensure that SCEs are in fact present in the population. For structural variants detected in single cells, detection of the same variant (on the same haplotype) in more than one cell can exclude BrdU incorporation as a possible cause.[17]
The number of single cell strands that need to be sequenced in order for an annotation to be accepted has yet to be proposed and is highly dependent on the questions being asked. As Strand-seq is founded on single cell sequencing techniques, one must consider the problems faced with single cell sequencing as well. These include the lacking standards for cell isolation and amplification. Even though previous Strand-seq studies isolated cells using FACS, microfluidics also serves as an attractive alternative. PCR has been shown to produce more erroneous amplification products compared to strand displacement based methods such as MDA and MALBAC, whereas the latter two techniques generate chimeric reads as a byproduct that can result in erroneous structural variation calls.[21][17] MDA and MALBAC also generate more dropouts than Strand-seq during SV detection because they require reads that cross the breakpoint of an SV to enable its detection (this is not required for any of the different SV classes that Strand-seq can detect).[17] Strand displacement amplification also tends to generate more sequence and longer products which could be beneficial for long read sequencing technologies.
References
- Falconer, Ester; Hills, Mark; Naumann, Ulrike; Poon, Steven S S; Chavez, Elizabeth A; Sanders, Ashley D; Zhao, Yongjun; Hirst, Martin; Lansdorp, Peter M (7 October 2012). "DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution". Nature Methods. 9 (11): 1107–1112. doi:10.1038/nmeth.2206. PMC 3580294. PMID 23042453.
- Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2002). Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-3218-3.
- Sotiropoulou, Panagiota A.; Candi, Aurélie; Blanpain, Cédric (November 2008). "The Majority of Multipotent Epidermal Stem Cells Do Not Protect Their Genome by Asymmetrical Chromosome Segregation". Stem Cells. 26 (11): 2964–2973. doi:10.1634/stemcells.2008-0634. PMID 18772311.
- Falconer, Ester; Chavez, Elizabeth; Henderson, Alexander; Lansdorp, Peter M (1 July 2010). "Chromosome orientation fluorescence in situ hybridization to study sister chromatid segregation in vivo". Nature Protocols. 5 (7): 1362–1377. doi:10.1038/nprot.2010.102. PMC 3771506. PMID 20595964.
- Li, Heng; Durbin, Richard (2009). "Fast and accurate short read alignment with Burrows–Wheeler transform". Bioinformatics. 25 (14): 1754–1760. doi:10.1093/bioinformatics/btp324. PMC 2705234. PMID 19451168.
- Langmead, Ben; Salzberg, Steven (2012). "Fast gapped-read alignment with Bowtie 2". Nature Methods. 9 (4): 357–359. doi:10.1038/nmeth.1923. PMC 3322381. PMID 22388286.
- Hills, Mark; o’Neill, Kieran; Falconer, Ester; Brinkman, Ryan; Lansdorp, Peter M (2013). "BAIT: Organizing genomes and mapping rearrangements in single cells". Genome Medicine. 5 (9): 82. doi:10.1186/gm486. PMC 3971352. PMID 24028793.
- Porubsky D, Ebert P, Audano PA, Vollger MR, Harvey WT, Marijon P, Ebler J, Munson KM, Sorensen M, Sulovari A, Haukness M, Ghareghani M, Lansdorp PM, Paten B, Devine SE, Sanders AD, Lee C, Chaisson MJ, Korbel JO, Eichler EE, Marschall T (March 2021). "Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads". Nat Biotechnol. 39 (3): 302–308. doi:10.1038/s41587-020-0719-5. PMC 7954704. PMID 33288906.
- Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, Sulovari A, Ebler J, Zhou W, Serra Mari R, Yilmaz F, Zhao X, Hsieh P, Lee J, Kumar S, Lin J, Rausch T, Chen Y, Ren J, Santamarina M, Höps W, Ashraf H, Chuang NT, Yang X, Munson KM, Lewis AP, Fairley S, Tallon LJ, Clarke WE, Basile AO, Byrska-Bishop M, Corvelo A, Evani US, Lu TY, Chaisson MJ, Chen J, Li C, Brand H, Wenger AM, Ghareghani M, Harvey WT, Raeder B, Hasenfeld P, Regier AA, Abel HJ, Hall IM, Flicek P, Stegle O, Gerstein MB, Tubio JM, Mu Z, Li YI, Shi X, Hastie AR, Ye K, Chong Z, Sanders AD, Zody MC, Talkowski ME, Mills RE, Devine SE, Lee C, Korbel JO, Marschall T, Eichler EE (April 2021). "Haplotype-resolved diverse human genomes and integrated analysis of structural variation". Science. 372 (6537). doi:10.1126/science.abf7117. PMC 8026704. PMID 33632895.
- Lansdorp, Eric; Linton, Lauren M.; Birren, Bruce; Nusbaum, Chad; Zody, Michael C.; Baldwin, Jennifer; Devon, Keri; Dewar, Ken; Doyle, Michael; Fitzhugh, William; Funke, Roel; Gage, Diane; Harris, Katrina; Heaford, Andrew; Howland, John; Kann, Lisa; Lehoczky, Jessica; Levine, Rosie; McEwan, Paul; McKernan, Kevin; Meldrim, James; Mesirov, Jill P.; Miranda, Cher; Morris, William; Naylor, Jerome; Raymond, Christina; Rosetti, Mark; Santos, Ralph; Sheridan, Andrew; et al. (2001). "Initial sequencing and analysis of the human genome" (PDF). Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011.
- Lark, K; Consigli, R; Minocha, H (1966). "Segregation of sister chromatids in mammalian cells". Science. 154 (3753): 1202–1205. Bibcode:1966Sci...154.1202L. doi:10.1126/science.154.3753.1202. PMID 5921385. S2CID 11039254.
- Rando, T (2007). "The Immortal Strand Hypothesis: Segregation and Reconstruction". Cell. 129 (7): 1239–1243. doi:10.1016/j.cell.2007.06.019. PMID 17604710.
- Falconer, Ester; Lansdorp, Peter M. (August 2013). "Strand-seq: A unifying tool for studies of chromosome segregation". Seminars in Cell & Developmental Biology. 24 (8–9): 643–652. doi:10.1016/j.semcdb.2013.04.005. PMC 3791154. PMID 23665005.
- Lansdorp, Peter (2007). "Immortal Strands? Give Me A Break". Cell. 129 (7): 1244–1247. doi:10.1016/j.cell.2007.06.017. PMID 17604711.
- Sanders AD, Hills M, Porubský D, Guryev V, Falconer E, Lansdorp PM (November 2016). "Characterizing polymorphic inversions in human genomes by single-cell sequencing". Genome Res. 26 (11): 1575–1587. doi:10.1101/gr.201160.115. PMC 5088599. PMID 27472961.
- Porubsky D, Sanders AD, Höps W, Hsieh P, Sulovari A, Li R, Mercuri L, Sorensen M, Murali SC, Gordon D, Cantsilieris S, Pollen AA, Ventura M, Antonacci F, Marschall T, Korbel JO, Eichler EE (August 2020). "Recurrent inversion toggling and great ape genome evolution". Nat Genet. 52 (8): 849–858. doi:10.1038/s41588-020-0646-x. PMC 7415573. PMID 32541924.
- Sanders AD, Meiers S, Ghareghani M, Porubsky D, Jeong H, van Vliet MA, Rausch T, Richter-Pechańska P, Kunz JB, Jenni S, Bolognini D, Longo GM, Raeder B, Kinanen V, Zimmermann J, Benes V, Schrappe M, Mardin BR, Kulozik AE, Bornhauser B, Bourquin JP, Marschall T, Korbel JO (March 2020). "Single-cell analysis of structural variations and complex rearrangements with tri-channel processing" (PDF). Nat Biotechnol. 38 (3): 343–354. doi:10.1038/s41587-019-0366-x. PMID 31873213. S2CID 209464011.
- Porubský D, Sanders AD, van Wietmarschen N, Falconer E, Hills M, Spierings DC, Bevova MR, Guryev V, Lansdorp PM (November 2016). "Direct chromosome-length haplotyping by single-cell sequencing". Genome Res. 26 (11): 1565–1574. doi:10.1101/gr.209841.116. PMC 5088598. PMID 27646535.
- LAMBERT, BO; HANSSON, KERSTIN; LINDSTEN, JAN; STEN, MARGARETA; WERELIUS, BARBRO (12 February 2009). "Bromodeoxyuridine-induced sister chromatid exchanges in human lymphocytes". Hereditas. 83 (2): 163–173. doi:10.1111/j.1601-5223.1976.tb01582.x. PMID 61954.
- Latt, Samuel (1974). "Sister chromatid exchanges, indices of human chromosome damage and repair: detection by fluorescence and induction by mitomycin C". Proceedings of the National Academy of Sciences of the United States of America. 71 (8): 3162–3166. Bibcode:1974PNAS...71.3162L. doi:10.1073/pnas.71.8.3162. PMC 388642. PMID 4137928.
- Blainey, Paul C. (May 2013). "The future is now: single-cell genomics of bacteria and archaea". FEMS Microbiology Reviews. 37 (3): 407–427. doi:10.1111/1574-6976.12015. PMC 3878092. PMID 23298390.
| Library resources about Single-cell DNA template strand sequencing |