Single-cell transcriptomics
Single-cell transcriptomics examines the gene expression level of individual cells in a given population by simultaneously measuring the RNA concentration (conventionally only messenger RNA (mRNA)) of hundreds to thousands of genes.[1] Single-cell transcriptomics makes it possible to unravel heterogeneous cell populations, reconstruct cellular developmental pathways, and model transcriptional dynamics — all previously masked in bulk RNA sequencing.[2]
Background
The development of high-throughput RNA sequencing (RNA-seq) and microarrays has made gene expression analysis a routine. RNA analysis was previously limited to tracing individual transcripts by Northern blots or quantitative PCR. Higher throughput and speed allow researchers to frequently characterize the expression profiles of populations of thousands of cells. The data from bulk assays has led to identifying genes differentially expressed in distinct cell populations, and biomarker discovery.[3]
These studies are limited as they provide measurements for whole tissues and, as a result, show an average expression profile for all the constituent cells. This has a couple of drawbacks. Firstly, different cell types within the same tissue can have distinct roles in multicellular organisms. They often form subpopulations with unique transcriptional profiles. Correlations in the gene expression of the subpopulations can often be missed due to the lack of subpopulation identification.[1] Secondly, bulk assays fail to recognize whether a change in the expression profile is due to a change in regulation or composition — for example if one cell type arises to dominate the population. Lastly, when your goal is to study cellular progression through differentiation, average expression profiles can only order cells by time rather than by developmental stage. Consequently, they cannot show trends in gene expression levels specific to certain stages.[4]
Recent advances in biotechnology allow the measurement of gene expression in hundreds to thousands of individual cells simultaneously. While these breakthroughs in transcriptomics technologies have enabled the generation of single-cell transcriptomic data, they also presented new computational and analytical challenges. Bioinformaticians can use techniques from bulk RNA-seq for single-cell data. Still, many new computational approaches have had to be designed for this data type to facilitate a complete and detailed study of single-cell expression profiles.[5]
Experimental steps
There is so far no standardized technique to generate single-cell data: all methods must include cell isolation from the population, lysate formation, amplification through reverse transcription and quantification of expression levels. Common techniques for measuring expression are quantitative PCR or RNA-seq.[6]
Isolating single cells
_B2.jpg.webp)
There are several methods available to isolate and amplify cells for single-cell analysis. Low throughput techniques are able to isolate hundreds of cells, are slow, and enable selection. These methods include:
- Micropipetting
- Cytoplasmic aspiration
- Laser capture microdissection.
High-throughput methods are able to quickly isolate hundreds to tens of thousands of cells.[7] Common techniques include:
- Fluorescence activated cell sorting (FACS)
- Microfluidic devices
Combining FACS with scRNA-seq has produced optimized protocols such as SORT-seq.[8] A list of studies that utilized SORT-seq can be found here.[9] Moreover, combining microfluidic devices with scRNA-seq has been optimized in 10x Genomics protocols.[10]
Quantitative PCR (qPCR)
To measure the level of expression of each transcript qPCR can be applied. Gene specific primers are used to amplify the corresponding gene as with regular PCR and as a result data is usually only obtained for sample sizes of less than 100 genes. The inclusion of housekeeping genes, whose expression should be constant under the conditions, is used for normalisation. The most commonly used house keeping genes include GAPDH and α-actin, although the reliability of normalisation through this process is questionable as there is evidence that the level of expression can vary significantly.[11] Fluorescent dyes are used as reporter molecules to detect the PCR product and monitor the progress of the amplification - the increase in fluorescence intensity is proportional to the amplicon concentration. A plot of fluorescence vs. cycle number is made and a threshold fluorescence level is used to find cycle number at which the plot reaches this value. The cycle number at this point is known as the threshold cycle (Ct) and is measured for each gene.[12]
Single-cell RNA-seq

The Single-cell RNA-seq technique converts a population of RNAs to a library of cDNA fragments. These fragments are sequenced by high-throughput next generation sequencing techniques and the reads are mapped back to the reference genome, providing a count of the number of reads associated with each gene.[13]
Normalisation of RNA-seq data accounts for cell to cell variation in the efficiencies of the cDNA library formation and sequencing. One method relies on the use of extrinsic RNA spike-ins (RNA sequences of known sequence and quantity) that are added in equal quantities to each cell lysate and used to normalise read count by the number of reads mapped to spike-in mRNA.[14]
Another control uses unique molecular identifiers (UMIs)-short DNA sequences (6–10nt) that are added to each cDNA before amplification and act as a bar code for each cDNA molecule. Normalisation is achieved by using the count number of unique UMIs associated with each gene to account for differences in amplification efficiency.[15]
A combination of both spike-ins, UMIs and other approaches have been combined for more accurate normalisation.
Considerations
A problem associated with single-cell data occurs in the form of zero inflated gene expression distributions, known as technical dropouts, that are common due to low mRNA concentrations of less-expressed genes that are not captured in the reverse transcription process. The percentage of mRNA molecules in the cell lysate that are detected is often only 10-20%.[16]
When using RNA spike-ins for normalisation the assumption is made that the amplification and sequencing efficiencies for the endogenous and spike-in RNA are the same. Evidence suggests that this is not the case given fundamental differences in size and features, such as the lack of a polyadenylated tail in spike-ins and therefore shorter length.[17] Additionally, normalisation using UMIs assumes the cDNA library is sequenced to saturation, which is not always the case.[15]
Data analysis
Insights based on single-cell data analysis assume that the input is a matrix of normalised gene expression counts, generated by the approaches outlined above, and can provide opportunities that are not obtainable by bulk.
Three main insights provided:[18]
- Identification and characterization of cell types and their spatial organisation in time
- Inference of gene regulatory networks and their strength across individual cells
- Classification of the stochastic component of transcription
The techniques outlined have been designed to help visualise and explore patterns in the data in order to facilitate the revelation of these three features.
Clustering
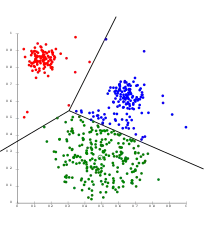

Clustering allows for the formation of subgroups in the cell population. Cells can be clustered by their transcriptomic profile in order to analyse the sub-population structure and identify rare cell types or cell subtypes. Alternatively, genes can be clustered by their expression states in order to identify covarying genes. A combination of both clustering approaches, known as biclustering, has been used to simultaneously cluster by genes and cells to find genes that behave similarly within cell clusters.[19]
Clustering methods applied can be K-means clustering, forming disjoint groups or Hierarchical clustering, forming nested partitions.
Biclustering
Biclustering provides several advantages by improving the resolution of clustering. Genes that are only informative to a subset of cells and are hence only expressed there can be identified through biclustering. Moreover, similarly behaving genes that differentiate one cell cluster from another can be identified using this method.[20]
Dimensionality reduction

Dimensionality reduction algorithms such as Principal component analysis (PCA) and t-SNE can be used to simplify data for visualisation and pattern detection by transforming cells from a high to a lower dimensional space. The result of this method produces graphs with each cell as a point in a 2-D or 3-D space. Dimensionality reduction is frequently used before clustering as cells in high dimensions can wrongly appear to be close due to distance metrics behaving non-intuitively.[21]
Principal component analysis
The most frequently used technique is PCA, which identifies the directions of largest variance principal components and transforms the data so that the first principal component has the largest possible variance, and successive principle components in turn each have the highest variance possible while remaining orthogonal to the preceding components. The contribution each gene makes to each component is used to infer which genes are contributing the most to variance in the population and are involved in differentiating different subpopulations.[22]
Differential expression
Detecting differences in gene expression level between two populations is used both single-cell and bulk transcriptomic data. Specialised methods have been designed for single-cell data that considers single cell features such as technical dropouts and shape of the distribution e.g. Bimodal vs. unimodal.[23]
Gene ontology enrichment
Gene ontology terms describe gene functions and the relationships between those functions into three classes:
- Molecular function
- Cellular component
- Biological process
Gene Ontology (GO) term enrichment is a technique used to identify which GO terms are over-represented or under-represented in a given set of genes. In single-cell analysis input list of genes of interest can be selected based on differentially expressed genes or groups of genes generated from biclustering. The number of genes annotated to a GO term in the input list is normalised against the number of genes annotated to a GO term in the background set of all genes in genome to determine statistical significance.[24]
Pseudotemporal ordering

Pseudo-temporal ordering (or trajectory inference) is a technique that aims to infer gene expression dynamics from snapshot single-cell data. The method tries to order the cells in such a way that similar cells are closely positioned to each other. This trajectory of cells can be linear, but can also bifurcate or follow more complex graph structures. The trajectory, therefore, enables the inference of gene expression dynamics and the ordering of cells by their progression through differentiation or response to external stimuli. The method relies on the assumptions that the cells follow the same path through the process of interest and that their transcriptional state correlates to their progression. The algorithm can be applied to both mixed populations and temporal samples.
More than 50 methods for pseudo-temporal ordering have been developed, and each has its own requirements for prior information (such as starting cells or time course data), detectable topologies, and methodology.[25] An example algorithm is the Monocle algorithm[26] that carries out dimensionality reduction of the data, builds a minimal spanning tree using the transformed data, orders cells in pseudo-time by following the longest connected path of the tree and consequently labels cells by type. Another example is the diffusion pseudotime (DPT) algorithm,[24] which uses a diffusion map and diffusion process. Another class of methods such as MARGARET [27] employ graph partitioning for capturing complex trajectory topologies such as disconnected and multifurcating trajectories.
Network inference
Gene regulatory network inference is a technique that aims to construct a network, shown as a graph, in which the nodes represent the genes and edges indicate co-regulatory interactions. The method relies on the assumption that a strong statistical relationship between the expression of genes is an indication of a potential functional relationship.[28] The most commonly used method to measure the strength of a statistical relationship is correlation. However, correlation fails to identify non-linear relationships and mutual information is used as an alternative. Gene clusters linked in a network signify genes that undergo coordinated changes in expression.[29]
Integration
The presence or strength of technical effects and the types of cells observed often differ in single-cell transcriptomics datasets generated using different experimental protocols and under different conditions. This difference results in strong batch effects that may bias the findings of statistical methods applied across batches, particularly in the presence of confounding.[30] As a result of the aforementioned properties of single-cell transcriptomic data, batch correction methods developed for bulk sequencing data were observed to perform poorly. Consequently, researchers developed statistical methods to correct for batch effects that are robust to the properties of single-cell transcriptomic data to integrate data from different sources or experimental batches. Laleh Haghverdi performed foundational work in formulating the use of mutual nearest neighbors between each batch to define batch correction vectors.[31] With these vectors, you can merge datasets that each include at least one shared cell type. An orthogonal approach involves the projection of each dataset onto a shared low-dimensional space using canonical correlation analysis.[32] Mutual nearest neighbors and canonical correlation analysis have also been combined to define integration "anchors" comprising reference cells in one dataset, to which query cells in another dataset are normalized.[33]
References
- Kanter, Itamar; Kalisky, Tomer (2015). "Single cell transcriptomics: methods and applications". Frontiers in Oncology. 5: 53. doi:10.3389/fonc.2015.00053. ISSN 2234-943X. PMC 4354386. PMID 25806353.
- Liu, Serena; Trapnell, Cole (2016). "Single-cell transcriptome sequencing: recent advances and remaining challenges". F1000Research. 5: F1000 Faculty Rev–182. doi:10.12688/f1000research.7223.1. ISSN 2046-1402. PMC 4758375. PMID 26949524.
- Szabo, David T. (2014). "Chapter 62 - Transcriptomic biomarkers in safety and risk assessment of chemicals". Biomarkers in Toxicology. Academic Press. pp. 1033–1038. ISBN 9780124046306.
- Trapnell, Cole (October 2015). "Defining cell types and states with single-cell genomics". Genome Research. 25 (10): 1491–1498. doi:10.1101/gr.190595.115. ISSN 1549-5469. PMC 4579334. PMID 26430159.
- Stegle, O.; Teichmann, S.; Marioni, J. (2015). "Computational and analytical challenges in single-cell transcriptomics". Nature Reviews Genetics. 16 (3): 133–145. doi:10.1038/nrg3833. PMID 25628217. S2CID 205486032.
- Kolodziejczyk, Aleksandra A.; Kim, Jong Kyoung; Svensson, Valentine; Marioni, John C.; Teichmann, Sarah A. (May 2015). "The Technology and Biology of Single-Cell RNA Sequencing". Molecular Cell. 58 (4): 610–620. doi:10.1016/j.molcel.2015.04.005. PMID 26000846.
- Poulin, Jean-Francois; Tasic, Bosiljka; Hjerling-Leffler, Jens; Trimarchi, Jeffrey M.; Awatramani, Rajeshwar (1 September 2016). "Disentangling neural cell diversity using single-cell transcriptomics". Nature Neuroscience. 19 (9): 1131–1141. doi:10.1038/nn.4366. ISSN 1097-6256. PMID 27571192. S2CID 14461377.
- Muraro, Mauro J.; Dharmadhikari, Gitanjali; Grün, Dominic; Groen, Nathalie; Dielen, Tim; Jansen, Erik; van Gurp, Leon; Engelse, Marten A.; Carlotti, Francoise; de Koning, Eelco J. P.; van Oudenaarden, Alexander (2016-10-26). "A Single-Cell Transcriptome Atlas of the Human Pancreas". Cell Systems. 3 (4): 385–394.e3. doi:10.1016/j.cels.2016.09.002. ISSN 2405-4712. PMC 5092539. PMID 27693023.
- "SORT-seq Archives". Single Cell Discoveries. Retrieved 2022-11-15.
- Zheng, Grace X. Y.; Terry, Jessica M.; Belgrader, Phillip; Ryvkin, Paul; Bent, Zachary W.; Wilson, Ryan; Ziraldo, Solongo B.; Wheeler, Tobias D.; McDermott, Geoff P.; Zhu, Junjie; Gregory, Mark T.; Shuga, Joe; Montesclaros, Luz; Underwood, Jason G.; Masquelier, Donald A. (2017-01-16). "Massively parallel digital transcriptional profiling of single cells". Nature Communications. 8 (1): 14049. Bibcode:2017NatCo...814049Z. doi:10.1038/ncomms14049. ISSN 2041-1723. PMC 5241818. PMID 28091601.
- Radonić, Aleksandar; Thulke, Stefanie; Mackay, Ian M.; Landt, Olfert; Siegert, Wolfgang; Nitsche, Andreas (23 January 2004). "Guideline to reference gene selection for quantitative real-time PCR". Biochemical and Biophysical Research Communications. 313 (4): 856–862. doi:10.1016/j.bbrc.2003.11.177. ISSN 0006-291X. PMID 14706621.
- Wildsmith, S. E.; Archer, G. E.; Winkley, A. J.; Lane, P. W.; Bugelski, P. J. (1 January 2001). "Maximization of signal derived from cDNA microarrays". BioTechniques. 30 (1): 202–206, 208. doi:10.2144/01301dd04. ISSN 0736-6205. PMID 11196312.
- Wang, Zhong; Gerstein, Mark; Snyder, Michael (23 March 2017). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews. Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. ISSN 1471-0056. PMC 2949280. PMID 19015660.
- Jiang, Lichun; Schlesinger, Felix; Davis, Carrie A.; Zhang, Yu; Li, Renhua; Salit, Marc; Gingeras, Thomas R.; Oliver, Brian (23 March 2017). "Synthetic spike-in standards for RNA-seq experiments". Genome Research. 21 (9): 1543–1551. doi:10.1101/gr.121095.111. ISSN 1088-9051. PMC 3166838. PMID 21816910.
- Islam, Saiful; Zeisel, Amit; Joost, Simon; La Manno, Gioele; Zajac, Pawel; Kasper, Maria; Lönnerberg, Peter; Linnarsson, Sten (1 February 2014). "Quantitative single-cell RNA-seq with unique molecular identifiers". Nature Methods. 11 (2): 163–166. doi:10.1038/nmeth.2772. ISSN 1548-7091. PMID 24363023. S2CID 6765530.
- Kharchenko, Peter V.; Silberstein, Lev; Scadden, David T. (1 July 2014). "Bayesian approach to single-cell differential expression analysis". Nature Methods. 11 (7): 740–742. doi:10.1038/nmeth.2967. ISSN 1548-7091. PMC 4112276. PMID 24836921.
- Svensson, Valentine; Natarajan, Kedar Nath; Ly, Lam-Ha; Miragaia, Ricardo J.; Labalette, Charlotte; Macaulay, Iain C.; Cvejic, Ana; Teichmann, Sarah A. (6 March 2017). "Power analysis of single-cell RNA-sequencing experiments". Nature Methods. advance online publication (4): 381–387. doi:10.1038/nmeth.4220. ISSN 1548-7105. PMC 5376499. PMID 28263961.
- Stegle, Oliver; Teichmann, Sarah A.; Marioni, John C. (1 March 2015). "Computational and analytical challenges in single-cell transcriptomics". Nature Reviews Genetics. 16 (3): 133–145. doi:10.1038/nrg3833. ISSN 1471-0056. PMID 25628217. S2CID 205486032.
- Buettner, Florian; Natarajan, Kedar N.; Casale, F. Paolo; Proserpio, Valentina; Scialdone, Antonio; Theis, Fabian J.; Teichmann, Sarah A.; Marioni, John C.; Stegle, Oliver (1 February 2015). "Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells". Nature Biotechnology. 33 (2): 155–160. doi:10.1038/nbt.3102. ISSN 1087-0156. PMID 25599176.
- Ntranos, Vasilis; Kamath, Govinda M.; Zhang, Jesse M.; Pachter, Lior; Tse, David N. (26 May 2016). "Fast and accurate single-cell RNA-seq analysis by clustering of transcript-compatibility counts". Genome Biology. 17 (1): 112. doi:10.1186/s13059-016-0970-8. ISSN 1474-7596. PMC 4881296. PMID 27230763.
- Pierson, Emma; Yau, Christopher (1 January 2015). "ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis". Genome Biology. 16: 241. doi:10.1186/s13059-015-0805-z. ISSN 1474-760X. PMC 4630968. PMID 26527291.
- Treutlein, Barbara; Brownfield, Doug G.; Wu, Angela R.; Neff, Norma F.; Mantalas, Gary L.; Espinoza, F. Hernan; Desai, Tushar J.; Krasnow, Mark A.; Quake, Stephen R. (15 May 2014). "Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq". Nature. 509 (7500): 371–375. Bibcode:2014Natur.509..371T. doi:10.1038/nature13173. PMC 4145853. PMID 24739965.
- Korthauer, Keegan D.; Chu, Li-Fang; Newton, Michael A.; Li, Yuan; Thomson, James; Stewart, Ron; Kendziorski, Christina (1 January 2016). "A statistical approach for identifying differential distributions in single-cell RNA-seq experiments". Genome Biology. 17 (1): 222. doi:10.1186/s13059-016-1077-y. ISSN 1474-760X. PMC 5080738. PMID 27782827.
- Haghverdi, Laleh; Büttner, Maren; Wolf, F. Alexander; Buettner, Florian; Theis, Fabian J. (1 October 2016). "Diffusion pseudotime robustly reconstructs lineage branching" (PDF). Nature Methods. 13 (10): 845–848. doi:10.1038/nmeth.3971. ISSN 1548-7091. PMID 27571553. S2CID 3594049.
- Saelens, Wouter; Cannoodt, Robrecht; Todorov, Helena; Saeys, Yvan (2018-03-05). "A comparison of single-cell trajectory inference methods: towards more accurate and robust tools". bioRxiv: 276907. doi:10.1101/276907. Retrieved 2018-03-12.
- Trapnell, Cole; Cacchiarelli, Davide; Grimsby, Jonna; Pokharel, Prapti; Li, Shuqiang; Morse, Michael; Lennon, Niall J.; Livak, Kenneth J.; Mikkelsen, Tarjei S.; Rinn, John L. (23 March 2017). "Pseudo-temporal ordering of individual cells reveals dynamics and regulators of cell fate decisions". Nature Biotechnology. 32 (4): 381–386. doi:10.1038/nbt.2859. ISSN 1087-0156. PMC 4122333. PMID 24658644.
- Pandey, Kushagra; Zafar, Hamim (2022). "Inference of cell state transitions and cell fate plasticity from single-cell with MARGARET". Nucleic Acids Research. 50 (15): e86. doi:10.1093/nar/gkac412. ISSN 0305-1048. PMC 9410915. PMID 35639499.
- Wei, J.; Hu, X.; Zou, X.; Tian, T. (1 December 2016). "Inference of genetic regulatory network for stem cell using single cells expression data". 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). pp. 217–222. doi:10.1109/BIBM.2016.7822521. ISBN 978-1-5090-1611-2. S2CID 27737735.
- Moignard, Victoria; Macaulay, Iain C.; Swiers, Gemma; Buettner, Florian; Schütte, Judith; Calero-Nieto, Fernando J.; Kinston, Sarah; Joshi, Anagha; Hannah, Rebecca; Theis, Fabian J.; Jacobsen, Sten Eirik; de Bruijn, Marella F.; Göttgens, Berthold (1 April 2013). "Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis". Nature Cell Biology. 15 (4): 363–372. doi:10.1038/ncb2709. ISSN 1465-7392. PMC 3796878. PMID 23524953.
- Hicks, Stephanie C; Townes, William F; Teng, Mingxiang; Irizarry, Rafael A (6 November 2017). "Missing data and technical variability in single-cell RNA-sequencing experiments". Biostatistics. 19 (4): 562–578. doi:10.1093/biostatistics/kxx053. PMC 6215955. PMID 29121214.
- Haghverdi, Laleh; Lun, Aaron T L; Morgan, Michael D; Marioni, John C (2 April 2018). "Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors". Nature Biotechnology. 36 (5): 421–427. doi:10.1038/nbt.4091. PMC 6152897. PMID 29608177.
- Butler, Andrew; Hoffman, Paul; Smibert, Peter; Papalexi, Efthymia; Satija, Rahul (2 April 2018). "Integrating single-cell transcriptomic data across different conditions, technologies, and species". Nature Biotechnology. 36 (5): 421–427. doi:10.1038/nbt.4096. PMC 6700744. PMID 29608179.
- Stuart, Tim; Butler, Andrew; Hoffman, Paul; Hafemeister, Christoph; Papalexia, Efthymia; Mauck, William M III; Hao, Yuhan; Marlon, Stoeckius; Smibert, Peter; Satija, Rahul (6 June 2019). "Comprehensive Integration of Single-Cell Data". Cell. 177 (7): 1888–1902. doi:10.1016/j.cell.2019.05.031. PMC 6687398. PMID 31178118.
External links
- Dissecting Tumor Heterogeneity with Single-Cell Transcriptomics
- The ultimate single-cell RNA sequencing guide by single-cell RNA sequencing service provider Single Cell Discoveries.