Single cell epigenomics
Single cell epigenomics is the study of epigenomics (the complete set of epigenetic modifications on the genetic material of a cell) in individual cells by single cell sequencing.[2][1][3] Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.

Single-cell DNA methylome sequencing
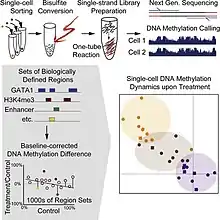
Single cell DNA genome sequencing quantifies DNA methylation. This is similar to single cell genome sequencing, but with the addition of a bisulfite treatment before sequencing. Forms include whole genome bisulfite sequencing,[4][5] and reduced representation bisulfite sequencing[6][7]

Single-cell ATAC-seq

ATAC-seq stands for Assay for Transposase-Accessible Chromatin with high throughput sequencing.[9] It is a technique used in molecular biology to identify accessible DNA regions, equivalent to DNase I hypersensitive sites.[9] Single cell ATAC-seq has been performed since 2015, using methods ranging from FACS sorting, microfluidic isolation of single cells, to combinatorial indexing.[8] In initial studies, the method was able to reliably separate cells based on their cell types, uncover sources of cell-to-cell variability, and show a link between chromatin organization and cell-to-cell variation.[8]
Single-cell ChIP-seq
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA.[9] ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins.[9] In epigenomics, this is often used to assess histone modifications (such as methylation).[9] ChIP-seq is also often used to determine transcription factor binding sites.[9]
Single-cell ChIP-seq is extremely challenging due to background noise caused by nonspecific antibody pull-down,[1] and only one study so far has performed it successfully. This study used a droplet-based microfluidics approach, and the low coverage required thousands of cells to be sequenced in order to assess cellular heterogeneity.[10][1]
Single-cell Hi-C
Chromosome conformation capture techniques (often abbreviated to 3C technologies or 3C-based methods[11]) are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in three dimensional space, even if the loci are separated by many kilobases[12] in the linear genome.
Currently, 3C methods start with a similar set of steps, performed on a sample of cells.[11] First, the cells are cross-linked, which introduces bonds between proteins, and between proteins and nucleic acids,[12] that effectively "freeze" interactions between genomic loci.[11] The genome is then cut digested into fragments through the use of restriction enzymes. Next, proximity based ligation is performed, creating long regions of hybrid DNA.[11] Lastly, the hybrid DNA is sequenced to determine genomic loci that are in close proximity to each other.[11]
Single-cell Hi-C is a modification of the original Hi-C protocol, which is an adaptation of the 3C method, that allows you to determine proximity of different regions of the genome in a single cell.[13] This method was made possible by performing the digestion and ligation steps in individual nuclei,[13] as opposed to the original Hi-C protocol, where ligation was performed after cell lysis in a pool containing crosslinked chromatin complexes.[14] In single cell Hi-C, after ligation, single cells are isolated and the remaining steps are performed in separate compartments,[13][15] and hybrid DNA is tagged with a compartment specific barcode. High-throughput sequencing is then performed on the pool of the hybrid DNA from the single cells. Although the recovery rate of sequenced interactions (hybrid DNA) can be as low as 2.5% of potential interactions,[16] it has been possible to generate three dimensional maps of entire genomes using this method.[17][18] Additionally, advances have been made in the analysis of Hi-C data, allowing for the enhancement of HiC datasets to generate even more accurate and detailed contact maps and 3D models.[15]
References
- Clark, Stephen J.; Lee, Heather J.; Smallwood, Sébastien A.; Kelsey, Gavin; Reik, Wolf (18 April 2016). "Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity". Genome Biology. 17 (1): 72. doi:10.1186/s13059-016-0944-x. PMC 4834828. PMID 27091476.
- Schwartzman, Omer; Tanay, Amos (13 October 2015). "Single-cell epigenomics: techniques and emerging applications". Nature Reviews Genetics. 16 (12): 716–726. doi:10.1038/nrg3980. PMID 26460349. S2CID 10326803.
- Hyun, Byung-Ryool; McElwee, John L.; Soloway, Paul D. (January 2015). "Single molecule and single cell epigenomics". Methods. 72: 41–50. doi:10.1016/j.ymeth.2014.08.015. PMC 4300266. PMID 25204781.
- Farlik, M; Sheffield, NC; Nuzzo, A; Datlinger, P; Schönegger, A; Klughammer, J; Bock, C (3 March 2015). "Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics". Cell Reports. 10 (8): 1386–97. doi:10.1016/j.celrep.2015.02.001. PMC 4542311. PMID 25732828.
- Smallwood, SA; Lee, HJ; Angermueller, C; Krueger, F; Saadeh, H; Peat, J; Andrews, SR; Stegle, O; Reik, W; Kelsey, G (August 2014). "Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity". Nature Methods. 11 (8): 817–20. doi:10.1038/nmeth.3035. PMC 4117646. PMID 25042786.
- Guo, H; Zhu, P; Wu, X; Li, X; Wen, L; Tang, F (December 2013). "Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing". Genome Research. 23 (12): 2126–35. doi:10.1101/gr.161679.113. PMC 3847781. PMID 24179143.
- Guo, H; Zhu, P; Guo, F; Li, X; Wu, X; Fan, X; Wen, L; Tang, F (May 2015). "Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing". Nature Protocols. 10 (5): 645–59. doi:10.1038/nprot.2015.039. PMID 25837417. S2CID 19510054.
- Pott, Sebastian; Lieb, Jason D. (21 August 2015). "Single-cell ATAC-seq: strength in numbers". Genome Biology. 16 (1): 172. doi:10.1186/s13059-015-0737-7. PMC 4546161. PMID 26294014.
- Meyer, Clifford A.; Liu, X. Shirley (16 September 2014). "Identifying and mitigating bias in next-generation sequencing methods for chromatin biology". Nature Reviews Genetics. 15 (11): 709–721. doi:10.1038/nrg3788. PMC 4473780. PMID 25223782.
- Rotem, A; Ram, O; Shoresh, N; Sperling, RA; Goren, A; Weitz, DA; Bernstein, BE (November 2015). "Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state". Nature Biotechnology. 33 (11): 1165–72. doi:10.1038/nbt.3383. PMC 4636926. PMID 26458175.
- de Wit, E.; de Laat, W. (3 January 2012). "A decade of 3C technologies: insights into nuclear organization". Genes & Development. 26 (1): 11–24. doi:10.1101/gad.179804.111. PMC 3258961. PMID 22215806.
- Mifsud, Borbala; Tavares-Cadete, Filipe; Young, Alice N.; Sugar, Robert; Schoenfelder, Stefan; Ferreira, Lauren; Wingett, Steven W.; Andrews, Simon; Grey, William; Ewels, Philip A.; Herman, Bram (June 2015). "Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C". Nature Genetics. 47 (6): 598–606. doi:10.1038/ng.3286. ISSN 1546-1718. PMID 25938943. S2CID 6036717.
- Nagano, Takashi; Lubling, Yaniv; Stevens, Tim J.; Schoenfelder, Stefan; Yaffe, Eitan; Dean, Wendy; Laue, Ernest D.; Tanay, Amos; Fraser, Peter (October 2013). "Single-cell Hi-C reveals cell-to-cell variability in chromosome structure". Nature. 502 (7469): 59–64. Bibcode:2013Natur.502...59N. doi:10.1038/nature12593. ISSN 0028-0836. PMC 3869051. PMID 24067610.
- Lieberman-Aiden, E.; van Berkum, N. L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B. R.; Sabo, P. J.; Dorschner, M. O.; Sandstrom, R. (2009-10-08). "Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome". Science. 326 (5950): 289–293. Bibcode:2009Sci...326..289L. doi:10.1126/science.1181369. ISSN 0036-8075. PMC 2858594. PMID 19815776.
- Zhang, Yan; An, Lin; Xu, Jie; Zhang, Bo; Zheng, W. Jim; Hu, Ming; Tang, Jijun; Yue, Feng (2018-02-21). "Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus". Nature Communications. 9 (1): 750. Bibcode:2018NatCo...9..750Z. doi:10.1038/s41467-018-03113-2. ISSN 2041-1723. PMC 5821732. PMID 29467363.
- Sekelja, Monika; Paulsen, Jonas; Collas, Philippe (7 April 2016). "4D nucleomes in single cells: what can computational modeling reveal about spatial chromatin conformation?". Genome Biology. 17 (1): 54. doi:10.1186/s13059-016-0923-2. PMC 4823877. PMID 27052789.
- Nagano, Takashi; Lubling, Yaniv; Varnai, Csilla; Dudley, Carmel; Leung, Wing; Baran, Yael; Mandelson-Cohen, Netta; Wingett, Steven; Fraser, Peter; Tanay, Amos (2016-12-15). "Cell cycle dynamics of chromosomal organisation at single-cell resolution". bioRxiv: 094466. doi:10.1101/094466.
- Stevens, Tim J.; Lando, David; Basu, Srinjan; Atkinson, Liam P.; Cao, Yang; Lee, Steven F.; Leeb, Martin; Wohlfahrt, Kai J.; Boucher, Wayne; O’Shaughnessy-Kirwan, Aoife; Cramard, Julie; Faure, Andre J.; Ralser, Meryem; Blanco, Enrique; Morey, Lluis; Sansó, Miriam; Palayret, Matthieu G. S.; Lehner, Ben; Di Croce, Luciano; Wutz, Anton; Hendrich, Brian; Klenerman, Dave; Laue, Ernest D. (13 March 2017). "3D structures of individual mammalian genomes studied by single-cell Hi-C". Nature. 544 (7648): 59–64. Bibcode:2017Natur.544...59S. doi:10.1038/nature21429. PMC 5385134. PMID 28289288.