Rosai–Dorfman disease
Rosai–Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy or sometimes as Destombes–Rosai–Dorfman disease,[1][2][3] is a rare disorder of unknown cause that is characterized by abundant histiocytes in the lymph nodes or other locations throughout the body.[1][4]
| Rosai-Dorfman disease | |
|---|---|
| Other names | Destombes-Rosaï-Dorfman disease |
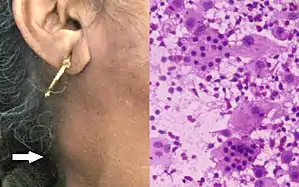 | |
| Patient with enlarged lymph node on neck caused by Rosai-Dorfman disease; H&E stain of fine-needle aspiration specimens showed histiocytes with intact lymphocytes and plasma cells inside them (i.e., emperipolesis). | |
| Specialty | Hematology |
Signs and symptoms
The histiocytosis of Destombes–Rosai–Dorfman disease can occur in lymph nodes, causing lymphadenopathy, or can occur outside lymph nodes in extranodal disease.
Lymphadenopathy
Lymphadenopathy can occur in one or more groups of lymph nodes. Among 358 cases of Destombes–Rosai–Dorfman disease that Rosai collected in a disease registry for which the location of lymphadenopathy was specified, 87.3% had cervical lymphadenopathy.[5] Axillary, inguinal, and mediastinal lymphadenopathy are also found in Destombes–Rosai–Dorfman disease.[5]
Extranodal disease
Accumulation of histiocytes may occur outside of lymph nodes. The most common sites of extranodal disease in Rosai's registry were skin, nasal cavity/paranasal sinuses, soft tissue, eyelid/orbit, bone, salivary glands, central nervous system, and heart.[6][5]
The symptoms of this disease vary with the site of accumulation similar to other regional tumors. For instance, accumulation in closed spaces such as the cranium can lead to poor outcomes compared to growth in the dermis of an extremity where surgical excision is possible.
Cause
The etiology of the condition is unknown. Possible but unproven infectious causes are Klebsiella, polyomaviridae, Epstein–Barr virus, parvovirus B19, and human herpesvirus 6.[1][7] Jilin University researchers suggested in 2017 that monocytes recruited to inflammatory lesions could produce macrophage colony-stimulating factor, which leads to a complex signal transduction, which leads to the histiocytosis characteristic of Destombes-Rosai–Dorfman disease.[7]
Diagnosis
The differential diagnosis of Destombes–Rosai–Dorfman disease includes both malignant and nonmalignant diseases, such as granulomatosis with polyangiitis, Langerhans cell histiocytosis, Langerhans cell sarcoma, lymphoma, sarcoidosis, IgG4-related disease, and tuberculosis.[1] The disease is diagnosed by biopsy of affected tissues. Microscopic examination of stained specimens will show histiocytes with lymphocytes and possibly other types of cells trapped within them, a phenomenon known as emperipolesis.[1][7] Upon immunohistochemical staining, the histiocytes will be positive for S100, CD68, and CD163 but negative for CD1a.[1][7]
Classification
In 2016 the Histiocyte Society proposed a classification of histiocytoses into five groups designated by letters: "C", "H", "L", "M", and "R".[8] Group "R" included Rosai–Dorfman disease and "miscellaneous noncutaneous, non-Langerhans cell histiocytoses".[8] Rosai–Dorfman disease itself was classified into "Familial", "Classical (nodal)", "Extranodal", "Neoplasia-associated", and "Immune disease-associated" subtypes.[8]
Treatment
Some patients have no symptoms, spontaneous remission,[9] or a relapsing/remitting course, making it difficult to decide whether therapy is needed.[1] In 2002, authors from Sapienza University of Rome stated on the basis of a comprehensive literature review that "clinical observation without treatment is advisable when possible".[10]
Therapeutic options include surgery, radiation therapy, and chemotherapy. Surgery is used to remove single lymph nodes, central nervous system lesions, or localized cutaneous disease.[1] In 2014, Dalia and colleagues wrote that for patients with extensive or systemic Destombes–Rosai–Dorfman disease, "a standard of care has not been established" concerning radiotherapy and chemotherapy.[1]
History
In 1965, Pierre-Paul Louis Lucien Destombes had described, in French, four patients having "adenitis with lipid excess" which is recognized as the original description of the condition.[1][2][3] Therefore, the condition is sometimes called "Destombes-Rosai-Dorfman disease".[2]
Four years later, in 1969, pathologists Juan Rosai and Ronald Dorfman published a paper on "sinus histiocytosis with massive lymphadenopathy".[1][11] With the discoveries that the condition can occur outside the head/neck region and in tissues other than lymph nodes, the condition later became known as "Rosai-Dorfman disease".
See also
References
- Dalia S, Sagatys E, Sokol L, Kubal T (2014). "Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment" (PDF). Cancer Control. 21 (4): 322–327. doi:10.1177/107327481402100408. PMID 25310213. Archived from the original (PDF) on 2015-04-23. Retrieved 2017-09-10.
- Becker MR, Gaiser T, Middel P, Rompel R (2008). "Clinicopathologic challenge. Destombes-Rosai-Dorfman disease (DRDD) (sinushistiocytosis with massive lymphadenopathy)". Int J Dermatol. 47 (2): 125–127. doi:10.1111/j.1365-4632.2008.03376.x. PMID 18211480. S2CID 41243620. Retrieved 2017-09-10.
- Destombes P (1965). "Adénites avec surcharge lipidique, de l'enfant ou de l'adulte jeune, observées aux Antilles et au Mali (quatre observations)" [Adenitis with lipid excess, in children or young adults, seen in the Antilles or Mali (4 cases)]. Bull Soc Pathol Exot Filiales (in French). 58 (6): 1169–1175. PMID 5899730.
- Riyaz N, Khader A, Sarita S (2005). "Rosai Dorfman syndrome". Indian J Dermatol Venereol Leprol. 71 (5): 342–344. doi:10.4103/0378-6323.16786. PMID 16394460.
- Foucar E, Rosai J, Dorfman R (1990). "Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity". Semin Diagn Pathol. 7 (1): 19–73. PMID 2180012.
- Heidarian, A; Anwar, A; Haseeb, MA; Gupta, R (2017). "Extranodal Rosai-Dorfman disease arising in the heart: clinical course and review of literature". Cardiovascular Pathology. 31: 1–4. doi:10.1016/j.carpath.2017.06.010. PMID 28797681.
- Cai Y, Shi Z, Bai Y (2017). "Review of Rosai-Dorfman disease: new insights into the pathogenesis of this rare disorder". Acta Haematol. 138 (1): 14–23. doi:10.1159/000475588. PMID 28614806. S2CID 31349416.
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM (2016). "Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages". Blood. 127 (22): 2672–2681. doi:10.1182/blood-2016-01-690636. PMC 5161007. PMID 26966089.
- Sardana, D., Goyal, A. and Gauba, K., 2015. Sinus histiocytosis with massive lymphadenopathy: a “massive” misnomer. Diagnostic cytopathology, 43(4), pp.315-319.
- Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, Villivà N, Mandelli F (2002). "Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review". Am J Hematol. 69 (1): 67–71. doi:10.1002/ajh.10008. PMID 11835335. S2CID 25696348.
- Rosai J, Dorfman RF (1969). "Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity". Arch Pathol. 87 (1): 63–70. PMID 5782438.