Site-specific recombination
Site-specific recombination, also known as conservative site-specific recombination, is a type of genetic recombination in which DNA strand exchange takes place between segments possessing at least a certain degree of sequence homology.[1][2][3] Enzymes known as site-specific recombinases (SSRs) perform rearrangements of DNA segments by recognizing and binding to short, specific DNA sequences (sites), at which they cleave the DNA backbone, exchange the two DNA helices involved, and rejoin the DNA strands. In some cases the presence of a recombinase enzyme and the recombination sites is sufficient for the reaction to proceed; in other systems a number of accessory proteins and/or accessory sites are required. Many different genome modification strategies, among these recombinase-mediated cassette exchange (RMCE), an advanced approach for the targeted introduction of transcription units into predetermined genomic loci, rely on SSRs.
Site-specific recombination systems are highly specific, fast, and efficient, even when faced with complex eukaryotic genomes.[4] They are employed naturally in a variety of cellular processes, including bacterial genome replication, differentiation and pathogenesis, and movement of mobile genetic elements.[5] For the same reasons, they present a potential basis for the development of genetic engineering tools.[6]
Recombination sites are typically between 30 and 200 nucleotides in length and consist of two motifs with a partial inverted-repeat symmetry, to which the recombinase binds, and which flank a central crossover sequence at which the recombination takes place. The pairs of sites between which the recombination occurs are usually identical, but there are exceptions (e.g. attP and attB of λ integrase).[7]
Classification: tyrosine- vs. serine- recombinases
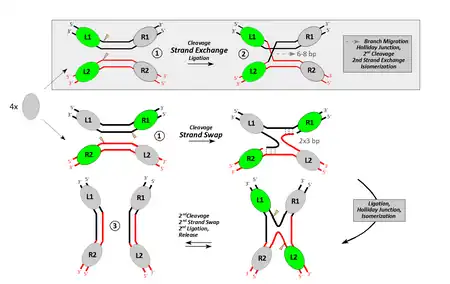
Top: Traditional view including strand-exchange followed by branch-migration (proofreading). The mechanism occurs in the framework of a synaptic complex (1) including both DNA sites in parallel orientation. While branch-migration explains the specific homology requirements and the reversibility of the process in a straightforward manner, it cannot be reconciled with the motions recombinase subunits have to undergo in three dimensions.
Bottom: Current view. Two simultaneous strand-swaps, each depending on the complementarity of three successive bases at (or close to) the edges of the 8-bp spacer (dashed lines indicate base-pairing). Didactic complications arise from the fact that, in this model, the synaptic complex must accommodate both substrates in an anti-parallel orientation.
This synaptic complex (1) arises from the association of two individual recombinase subunits ("protomers"; gray ovals) with the respective target site. Its formation depends on inter-protomer contacts and DNA bending, which in turn define the subunits (green) with an active role during the first crossover reaction. Both representations illustrate only one half of the respective pathway. These parts are separated by a Holliday junction/isomerization step before the product (3) can be released.
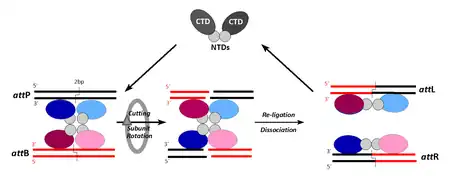
Contrary to Tyr-recombinases, the four participating DNA strands are cut in synchrony at points staggered by only 2 bp (leaving little room for proofreading). Subunit-rotation (180°) permits the exchange of strands while covalently linked to the protein partner. The intermediate exposure of double-strand breaks bears risks of triggering illegitimate recombination and thereby secondary reactions.
Here, the synaptic complex arises from the association of pre-formed recombinase dimers with the respective target sites (CTD/NTD, C-/N-terminal domain). Like for Tyr-recombinases, each site contains two arms, each accommodating one protomer. As both arms are structured slightly differently, the subunits become conformationally tuned and thereby prepared for their respective role in the recombination cycle. Contrary to members of the Tyr-class the recombination pathway converts two different substrate sites (attP and attB) to site-hybrids (attL and attR). This explains the irreversible nature of this particular recombination pathway, which can only be overcome by auxiliary "recombination directionality factors" (RDFs).
Based on amino acid sequence homologies and mechanistic relatedness, most site-specific recombinases are grouped into one of two families: the tyrosine (Tyr) recombinase family or serine (Ser) recombinase family. The names stem from the conserved nucleophilic amino acid residue present in each class of recombinase which is used to attack the DNA and which becomes covalently linked to it during strand exchange. The earliest identified members of the serine recombinase family were known as resolvases or DNA invertases, while the founding member of the tyrosine recombinases, lambda phage integrase (using attP/B recognition sites), differs from the now well-known enzymes such as Cre (from the P1 phage) and FLP (from the yeast Saccharomyces cerevisiae). Famous serine recombinases include enzymes such as gamma-delta resolvase (from the Tn1000 transposon), Tn3 resolvase (from the Tn3 transposon), and φC31 integrase (from the φC31 phage).[8]
Although the individual members of the two recombinase families can perform reactions with the same practical outcomes, the families are unrelated to each other, having different protein structures and reaction mechanisms. Unlike tyrosine recombinases, serine recombinases are highly modular, as was first hinted by biochemical studies[9] and later shown by crystallographic structures.[10][11] Knowledge of these protein structures could prove useful when attempting to re-engineer recombinase proteins as tools for genetic manipulation.
Mechanism
Recombination between two DNA sites begins by the recognition and binding of these sites – one site on each of two separate double-stranded DNA molecules, or at least two distant segments of the same molecule – by the recombinase enzyme. This is followed by synapsis, i.e. bringing the sites together to form the synaptic complex. It is within this synaptic complex that the strand exchange takes place, as the DNA is cleaved and rejoined by controlled transesterification reactions. During strand exchange, each double-stranded DNA molecule is cut at a fixed point within the crossover region of the recognition site, releasing a deoxyribose hydroxyl group, while the recombinase enzyme forms a transient covalent bond to a DNA backbone phosphate. This phosphodiester bond between the hydroxyl group of the nucleophilic serine or tyrosine residue conserves the energy that was expended in cleaving the DNA. Energy stored in this bond is subsequently used for the rejoining of the DNA to the corresponding deoxyribose hydroxyl group on the other DNA molecule. The entire reaction therefore proceeds without the need for external energy-rich cofactors such as ATP.
Although the basic chemical reaction is the same for both tyrosine and serine recombinases, there are some differences between them.[12] Tyrosine recombinases, such as Cre or FLP, cleave one DNA strand at a time at points that are staggered by 6–8bp, linking the 3’ end of the strand to the hydroxyl group of the tyrosine nucleophile (Fig. 1).[13] Strand exchange then proceeds via a crossed strand intermediate analogous to the Holliday junction in which only one pair of strands has been exchanged.[14][15]
The mechanism and control of serine recombinases is much less well understood. This group of enzymes was only discovered in the mid-1990s and is still relatively small. The now classical members gamma-delta and Tn3 resolvase, but also new additions like φC31-, Bxb1-, and R4 integrases, cut all four DNA strands simultaneously at points that are staggered by 2 bp (Fig. 2).[16] During cleavage, a protein–DNA bond is formed via a transesterification reaction, in which a phosphodiester bond is replaced by a phosphoserine bond between a 5’ phosphate at the cleavage site and the hydroxyl group of the conserved serine residue (S10 in resolvase).[17][18]


It is still not entirely clear how the strand exchange occurs after the DNA has been cleaved. However, it has been shown that the strands are exchanged while covalently linked to the protein, with a resulting net rotation of 180°.[19][20] The most quoted (but not the only) model accounting for these facts is the "subunit rotation model" (Fig. 2).[12][21] Independent of the model, DNA duplexes are situated outside of the protein complex, and large movement of the protein is needed to achieve the strand exchange. In this case the recombination sites are slightly asymmetric, which allows the enzyme to tell apart the left and right ends of the site. When generating products, left ends are always joined to the right ends of their partner sites, and vice versa. This causes different recombination hybrid sites to be reconstituted in the recombination products. Joining of left ends to left or right to right is avoided due to the asymmetric “overlap” sequence between the staggered points of top and bottom strand exchange, which is in stark contrast to the mechanism employed by tyrosine recombinases.[12]
The reaction catalysed by Cre-recombinase, for instance, may lead to excision of the DNA segment flanked by the two sites (Fig. 3A), but may also lead to integration or inversion of the orientation of the flanked DNA segment (Fig. 3B). What the outcome of the reaction will be is dictated mainly by the relative locations and orientations of the sites that are to be recombined, but also by the innate specificity of the site-specific system in question. Excisions and inversions occur if the recombination takes place between two sites that are found on the same molecule (intramolecular recombination), and if the sites are in the same (direct repeat) or in an opposite orientation (inverted repeat), respectively. Insertions, on the other hand, take place if the recombination occurs on sites that are situated on two different DNA molecules (intermolecular recombination), provided that at least one of these molecules is circular. Most site-specific systems are highly specialised, catalysing only one of these different types of reaction, and have evolved to ignore the sites that are in the "wrong" orientation.
See also
References
- Bode, J; Schlake, T; asadasasada Iber, M; Schuebeler, D; Seibler, J; Snezhkov, E; Nikolaev, L (2000). "The transgeneticist's toolbox: novel methods for the targeted modification of eukaryotic genomes". Biol. Chem. 381 (9–10): 801–813. doi:10.1515/BC.2000.103. PMID 11076013. S2CID 36479502.
- Kolb, A.F. (2002). "Genome Engineering Using Site-Specific Recombinases". Cloning & Stem Cells. 4 (1): 65–80. doi:10.1089/153623002753632066. PMID 12006158.
- Coates, C.J.; Kaminski, JM; Summers, JB; Segal, DJ; Miller, AD; Kolb, AF (2005). "Site-directed genome modification: derivatives of BAL-modifying enzymes as targeting tools" (PDF). Trends in Biotechnology. 23 (8): 407–19. doi:10.1016/j.tibtech.2005.06.009. PMID 15993503. Archived from the original (PDF) on 2006-08-29.
- Sauer, B. (1998). "Inducible Gene Targeting in Mice Using the Cre/loxSystem" (PDF). Methods. 14 (4): 381–92. doi:10.1006/meth.1998.0593. PMID 9608509. Archived from the original (PDF) on 2011-06-11.
- Nash, H. A. (1996). Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments. Escherichia coli and Salmonella: cellular and molecular biology, 2, pp. 2363–2376.
- Akopian, A.; Stark, W.M. (2005). Site-Specific DNA Recombinases as Instruments for Genomic Surgery. Advances in Genetics. Vol. 55. pp. 1–23. doi:10.1016/S0065-2660(05)55001-6. ISBN 978-0-12-017655-7. PMID 16291210.
- Landy, A. (1989). "Dynamic, Structural, and Regulatory Aspects of lambda Site-Specific Recombination". Annual Review of Biochemistry. 58 (1): 913–41. doi:10.1146/annurev.bi.58.070189.004405. PMID 2528323.
- Stark, W.M.; Boocock, M.R. (1995). "Topological selectivity in site-specific recombination". Mobile Genetic Elements. Oxford University Press. pp. 101–129.
- Abdel-Meguid, S.S.; Grindley, N.D.; Templeton, N.S.; Steitz, T.A. (April 1984). "Cleavage of the site-specific recombination protein gamma delta resolvase: the smaller of two fragments binds DNA specifically". Proc. Natl. Acad. Sci. U.S.A. 81 (7): 2001–5. Bibcode:1984PNAS...81.2001A. doi:10.1073/pnas.81.7.2001. PMC 345424. PMID 6326096.
- Yang, W.; Steitz, T.A. (1995). "Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site". Cell. 82 (2): 193–207. doi:10.1016/0092-8674(95)90307-0. PMID 7628011. S2CID 15849525.
- Li, W.; Kamtekar, S; Xiong, Y; Sarkis, GJ; Grindley, ND; Steitz, TA (2005). "Structure of a Synaptic Gamma Delta Resolvase Tetramer Covalently Linked to Two Cleaved DNAs". Science. 309 (5738): 1210–5. Bibcode:2005Sci...309.1210L. doi:10.1126/science.1112064. PMID 15994378. S2CID 84409916.
- Turan, S.; Bode, J. (2011). "Review: Site-specific recombinases: from tag-and-target- to tag-and-exchange-based genomic modifications". FASEB J. 25 (12): 4088–4107. doi:10.1096/fj.11-186940. PMID 21891781. S2CID 7075677.
- Van Duyne, G.D. (2002). "A structural view of tyrosine recombinase site-specific recombination". Mobile DNA II. ASM Press. pp. 93–117.
- Holliday, R. (1964). "A mechanism for gene conversion in fungi" (PDF). Genetics Research. 5 (2): 282–304. doi:10.1017/S0016672300001233.
- Grainge, I.; Jayaram, M. (1999). "The integrase family of recombinases: organization and function of the active site". Molecular Microbiology. 33 (3): 449–56. doi:10.1046/j.1365-2958.1999.01493.x. PMID 10577069.
- Stark, W.M.; Boocock, M.R.; Sherratt, DJ (1992). "Catalysis by site-specific recombinases". Trends in Genetics. 8 (12): 432–9. doi:10.1016/0168-9525(92)90327-Z. PMID 1337225.
- Reed, R.R.; Grindley, N.D. (1981). "Transposon-mediated site-specific recombination in vitro: DNA cleavage and protein-DNA linkage at the recombination site". Cell. 25 (3): 721–8. doi:10.1016/0092-8674(81)90179-3. PMID 6269756. S2CID 28410571.
- Reed, R.R.; Moser, C.D. (1984). "Resolvase-mediated recombination intermediates contain a serine residue covalently linked to DNA". Cold Spring Harb Symp Quant Biol. 49: 245–9. doi:10.1101/sqb.1984.049.01.028. PMID 6099239.
- Stark, M.W.; Sherratt, DJ; Boocock, MR (1989). "Site-specific recombination by Tn 3 resolvase: topological changes in the forward and reverse reactions". Cell. 58 (4): 779–90. doi:10.1016/0092-8674(89)90111-6. PMID 2548736. S2CID 46508016.
- Stark, W.M.; Boocock, M.R. (1994). "The Linkage Change of a Knotting Reaction Catalysed by Tn3 Resolvase". Journal of Molecular Biology. 239 (1): 25–36. doi:10.1006/jmbi.1994.1348. PMID 8196046.
- Sarkis, G.J; Murley, LL; Leschziner, AE; Boocock, MR; Stark, WM; Grindley, ND (2001). "A model for the gamma-delta resolvase synaptic complex". Molecular Cell. 8 (3): 623–31. doi:10.1016/S1097-2765(01)00334-3. PMID 11583624.