Smoker's macrophages
Smoker’s macrophages are alveolar macrophages whose characteristics, including appearance, cellularity, phenotypes, immune response, and other functions, have been affected upon the exposure to cigarettes.[1] These altered immune cells are derived from several signaling pathways and are able to induce numerous respiratory diseases. They are involved in asthma, chronic obstructive pulmonary diseases (COPD), pulmonary fibrosis, and lung cancer.[2] Smoker’s macrophages are observed in both firsthand and secondhand smokers, so anyone exposed to cigarette contents, or cigarette smoke extract (CSE), would be susceptible to these macrophages, thus in turns leading to future complications.[3]

Alveolar macrophages are crucial in processing inhaled substances including cigarette chemicals and particulate matter.[4] The chemicals in tobacco, such as nicotine, tar, and carbon monoxide, stimulate several physiological pathways, which influence the recruitment and functions of these macrophages. Some of the smoker’s macrophages are recruited from the circulating monocytes while some are the original alveolar macrophages residing in the lung. The biochemical processes also lead to immunomodulation and dysregulated repair processes, so the malfunction of macrophages renders individuals more susceptible to infections.[5][2] In addition, these inhaled substances can enter the bloodstream, especially nicotine which is rapidly transported to the brain, leading to addiction; it will subsequently distributed throughout the body, leading to carcinoma in the future.[6]

The morbidity of cigarette smoking is nearly 50% with 7 million first-hand smokers and 1.2 millions second hand smokers killed each year[8]. Regardless of active or passive smokers, macrophage accumulation is found in the lungs.[3][5] The diagnostic methods for smoke-related diseases include bronchoalveolar lavage which can also be used for examining smoker's macrophages in addition to augmented inflammatory cells in the alveolar lumen.[1]
Appearance
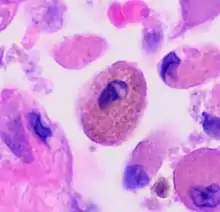
Autofluorescence
The uptake of tar from cigarettes accumulates in alveolar macrophages and causes autofluorescence.[4] The intensity of fluorescence, however, is independent of cigarette exposure. This indicates a maximum capacity of tar uptake; excess tar cannot be retained by macrophages. Another pigment in smoker’s macrophage is hemosiderin which is involved in iron homoeostasis. Hemosiderin-laden pigmented macrophages are yellowish brown and found in the bronchiole and peribronchiolar alveolar space.[9] The presence of these dirty macrophages has been a characteristic of many smoke-related lung diseases.[9][10]
Physiological pathways mediating macrophage changes
Macrophage Polarization
A macrophage can be polarized into the classic M1 or M2 phenotype, and this phenomenon can be seen in cigarette consumption.[2] In this polarization scheme, lower M1 markers and higher M2 markers have been observed.[4] The reprogram of macrophage implies a dysregulated inflammation that can damage healthy lung cells.
Macrophage polarization is mediated by three major signaling pathways: NF-κB, MAPK, and JAK/STAT.[2] Each signaling cascade can lead to different results depending on the length of smoke history. It is therefore important to evaluate the characteristics of research participants and specify the experimental conditions when examining smoker’s macrophages. It is anticipated that cigarette smoking inhibits signal transduction which alters gene expression and cytokine profile with increasing M2-like phenotype. This trait is involved in anti-inflammation and tissue repair, but this can also be pro-fibrotic.[11] However, some studies found variation in the conventional polarization and found dual polarization in multiple diseases, yet the direction and extent of polarization are also different across diseases.[4] Despite the contradiction, treatments targeting the polarization process have promising results.
NF-κB
In long-term smoking or established diseases, not only does CSE decrease the production of pro-inflammatory cytokines, but also impairs TLR2 and TLR4 signaling.[2] Its inhibitory effect on NF-κB also induces apoptosis of alveolar macrophage. Prolonged exposure to CSE hence leads to M2 polarization. Meanwhile, NF-κB pathways will be activated with low concentration of CSE or in previously unexposed individuals. The increased activity of NF-κB upregulates the production of pro-inflammatory cytokines TNF-α, IL-1β, and IL-8. The short-term exposure attracts macrophages and neutrophils to the lung with a 4-fold increase in cellularity.[4][2] Short duration also biases polarization towards M1 phenotype. The number of immune cells however will be normalized in 6 months, demonstrating the shift in signaling direction.
MAPK
Similar time and dose dependent effects of CSE are exerted on macrophage polarization through the MAPK signaling pathway which involves JNK and ERK as intermediate signaling molecules.[2] In diseased conditions due to long-term smoking, the inactivation of JNK reduces the levels of reactive nitrogen species and pro-inflammatory cytokines with more M2-like phenotype. However, brief exposure to CSE triggers the activation of ERK that increases MUC1, TNF-a, and IL-8 levels to produce inflammatory effects.
JAK/STAT
Cigarette contents also modulate multiple STAT proteins activities.[2] In response to the smoking, STAT3 and STAT6 signaling are stimulated to potentiate M2-like phenotype with elevated IL-6, IL-10, IL-12, and TGF-b. In the meantime, the toxic nitrogenous chemicals and oxidative stress would be reduced. In post-smoke situations, reduction in STAT1 is associated with M1-like phenotype and the downregulation of IFN-γ signaling.
Cholinergic Anti-inflammatory pathway
Nicotine in cigarettes modulates the above signaling pathways by binding to α-7 nicotinic receptors on macrophage or neurons, hence activating the cholinergic anti-inflammatory pathway.[12] Changes can thus be directly mediated by binding of nicotine to macrophage or indirectly via the Vagus nerve. Upon binding, the inhibition of the NF-κB and activation of JAK2/STAT3 pathways lead to over-inhibition of pro-inflammatory cytokines and thus an imbalance toward anti-inflammatory cytokines. The result may be lethal if inflammation is not controlled.
Function abnormalities
Iron homeostasis

Cigarettes contain a small amount of iron, but cumulatively a larger quantity in daily smoking.[13] The increasing iron exposure in the lung and airway affects both respiratory and systemic iron homeostasis by modifying cellular response. Although direct etiologic link has not been established, there is a 4-fold increase in intracellular iron level and a concomitant iron release observed in smoker’s macrophage.[4][13] While iron-loading affects macrophage activation and functions, excessive extracellular iron favors bacterial growth. Normally, activated alveolar macrophage secretes lipocalin-2 which traps bacterial siderophores and prevents bacterial iron uptake. Iron imbalance locally in the lung thus results in higher risk of infection.
Hemosiderin is the iron storage in smoker's macrophage rather than ferritin. It is formed during hemorrhage or abnormal metabolism of ferritin.[14][15] Indeed, buildup of iron causes oxidative stress resulting in lung damage and mitochondrial dysfunction.[13] The level of hemosiderin-laden macrophage is also associated with pulmonary hemodynamics parameters which is used to evaluate pulmonary hypertension in the early stage of diseases.[16]
Iron homeostasis has been associated with macrophage polarization and reprogramming despite unclear causality in cigarette iron.[13] M1 macrophage demonstrates high TF, HAMP, and FTH1 gene activities that mediates iron uptake. M2 macrophage on the other hand expresses FPN1 which causes iron release. Supplementing iron to mice predisposes macrophage to M2 phenotype and inhibits M1-mediated inflammation.
Immune Response
The immune functions in smoker’s macrophages are compromised, so the airway pathogens are more likely to accumulate and cause infection.[17] Smoker’s macrophages have reduced expression of HLA-DR antigens, causing immunosuppression.[18][19] In addition, nicotine impairs the phagocytosis of M. tuberculosis and also induces immunosuppression via the activation of alpha-7 nicotinic receptors.[4][17] Meanwhile, due to the impaired TLR2 and TLR4 signaling, macrophages fail to recognize pathogens, so there is a decrease in pathogen clearance.[17] Therefore, smokers are prone to acute respiratory tract infection and community acquired pneumonia.[20]
Disease phenotype
Asthma
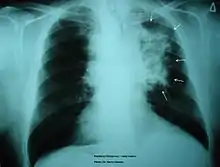
Asthma has been proven to have a causal relationship with smoking due to the modified inflammation reaction.[21] Alveolar macrophages will be excessively recruited onto the airway wall, leading to a narrower airway for oxygen to pass through. Some patients may also be affected by airway remodeling. Smoker's macrophages affect the elastic fibers in the mucus layer of the airways, tightening the lumen and causing asthma. Symptoms of asthma include wheezing, coughing, and chest discomfort. To ameliorate the situation, drugs that either suppress the inflammatory response or relaxes the airway will be administrated, so air can pass through.[22]
Chronic Obstructive Pulmonary Disease
Smoking is found to be the most important causative factor leading to COPD.[23] Because of the altered inflammatory response of the macrophages, smoking induces inflammation across the entire airway, which in turns obstructs the airflow. Symptoms of COPD include persistent coughing, wheezing, chest infections and breathlessness. Treatments for COPD usually focus on the source of the problem, which is smoking, thus the general treatment is going through smoking rehabilitation which including nicotine replacement therapy, mental therapy for advice, and support to quit smoking. In certain urgent cases, direct constriction also occurs, in which bronchodilators allow the airway to dilate.
Cancer
Tobacco smoking has been associated with cancer mainly along the respiratory tract, but may also lead to cancer in the bladder and renal pelvis.[24] Upon smoking, carcinogenic chemicals are inhaled, affecting the inflammation response. As inflammation plays an important role in inducing cancer, with smoking affecting the inflammatory response of macrophages in the lungs, the dysregulated inflammatory response poses a higher risk in developing cancer along the airway. Symptoms of cancer mediated by cancer include lumps on the body, sudden weight loss, persistent coughing and tough swallowing. Treatments for cancer are generally surgery, chemotherapy and radiation therapy. These treatments directly target the cancer cells to kill the cancer prior to smoking rehabilitation programs.
Prognosis
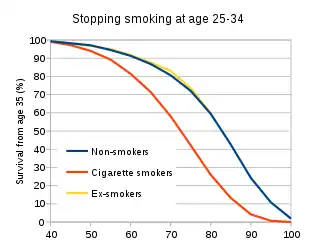
Smoking cessation is one the most effective methods for managing numerous smoke-related diseases and other immune diseases such as AIDs.[25][26] It brings both short term and long term benefits as the mucus clearance is improved in 48 hours and the mortality risk of lung cancer is halved in 10 years.[25] In addition, the immune system starts to recover in 15 days as the inhibitory effects of cigarettes on macrophages are removed.[27] The risks of morbidity and mortality of infectious diseases are significantly reduced in 1 year and become comparable with non-smokers after 5 years of quitting.[20] Meanwhile, the life expectancy after smoking cessation increased by 10 years with the reduced risks of these diseases.[28] Furthermore, early cessation in the age of 25-34 enhances the survival rate at the age of 35 by 20-30% compared with an average smoker.[29]
Future research
Cigarette smoking has been extensively researched to understand the mechanisms of how it affects the macrophages and causes diseases. Meanwhile, novel therapies are being developed to target the molecular pathways.[2] Future treatments have higher specificity and can potentially reverse the changes to macrophage and the cytokine profile, hence improving the clinical outcomes of related diseases. In the meantime, governments around the globe are encouraging smoking cessation and some progress is made.[30] However, the psychological challenge for smokers to commit to quitting is often omitted.[31] For instance, smoking cessation has been associated with depression while individuals may also experience withdrawal symptoms.[31][32] The psychological aspect of smoking could be further investigated to formulate a better rehabilitation strategy to aid smoking cessation.
References
- Monick, Martha M.; Powers, Linda S.; Walters, Katherine; Lovan, Nina; Zhang, Michael; Gerke, Alicia; Hansdottir, Sif; Hunninghake, Gary W. (2010-11-01). "Identification of an Autophagy Defect in Smokers' Alveolar Macrophages". The Journal of Immunology. 185 (9): 5425–5435. doi:10.4049/jimmunol.1001603. ISSN 0022-1767. PMC 3057181. PMID 20921532.
- Yang, David C.; Chen, Ching-Hsien (November 2018). "Cigarette Smoking-Mediated Macrophage Reprogramming: Mechanistic Insights and Therapeutic Implications". Journal of Nature and Science. 4 (11): e539. ISSN 2377-2700. PMC 6383770. PMID 30801020.
- Woodruff, Prescott G.; Ellwanger, Almut; Solon, Margaret; Cambier, Christopher J.; Pinkerton, Kent E.; Koth, Laura L. (April 2009). "Alveolar Macrophage Recruitment and Activation by Chronic Second Hand Smoke Exposure in Mice". COPD. 6 (2): 86–94. doi:10.1080/15412550902751738. ISSN 1541-2555. PMC 2873864. PMID 19378221.
- Lugg, Sebastian T.; Scott, Aaron; Parekh, Dhruv; Naidu, Babu; Thickett, David R. (2022-01-01). "Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease". Thorax. 77 (1): 94–101. doi:10.1136/thoraxjnl-2020-216296. ISSN 0040-6376. PMC 8685655. PMID 33986144.
- Wynn, Thomas A.; Vannella, Kevin M. (2016-03-15). "Macrophages in tissue repair, regeneration, and fibrosis". Immunity. 44 (3): 450–462. doi:10.1016/j.immuni.2016.02.015. ISSN 1074-7613. PMC 4794754. PMID 26982353.
- Tega, Yuma; Yamazaki, Yuhei; Akanuma, Shin-ichi; Kubo, Yoshiyuki; Hosoya, Ken-ichi (2018). "Impact of Nicotine Transport across the Blood–Brain Barrier: Carrier-Mediated Transport of Nicotine and Interaction with Central Nervous System Drugs". Biological and Pharmaceutical Bulletin. 41 (9): 1330–1336. doi:10.1248/bpb.b18-00134. PMID 30175770. S2CID 52143978.
- CDCTobaccoFree (2020-10-06). "Tobacco-Related Mortality". Centers for Disease Control and Prevention. Retrieved 2022-04-19.
- "Tobacco". www.who.int. Retrieved 2022-04-17.
- Rossi, Giulio; Cavazza, Alberto; Spagnolo, Paolo; Bellafiore, Salvatore; Kuhn, Elisabetta; Carassai, Pierpaolo; Caramanico, Laura; Montanari, Gloria; Cappiello, Gaia; Andreani, Alessandro; Bono, Francesca (2017-09-30). "The role of macrophages in interstitial lung diseases: Number 3 in the Series "Pathology for the clinician" Edited by Peter Dorfmüller and Alberto Cavazza". European Respiratory Review. 26 (145). doi:10.1183/16000617.0009-2017. ISSN 0905-9180. PMC 9488916. PMID 28724562. S2CID 23917003.
- Mukhopadhyay, Sanjay (2000). Non-Neoplastic Pulmonary Pathology: An Algorithmic Approach to Histologic Findings in the Lung. Cambridge: Cambridge University Press. doi:10.1017/cbo9781107445079. ISBN 9781107445079.
- Lis-López, Lluis; Bauset, Cristina; Seco-Cervera, Marta; Cosín-Roger, Jesús (2021-11-23). "Is the Macrophage Phenotype Determinant for Fibrosis Development?". Biomedicines. 9 (12): 1747. doi:10.3390/biomedicines9121747. ISSN 2227-9059. PMC 8698841. PMID 34944564.
- Báez-Pagán, Carlos A.; Delgado-Vélez, Manuel; Lasalde-Dominicci, José A. (September 2015). "Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation". Journal of Neuroimmune Pharmacology. 10 (3): 468–476. doi:10.1007/s11481-015-9601-5. ISSN 1557-1890. PMC 4546521. PMID 25870122.
- Zhang, William Z.; Butler, James J.; Cloonan, Suzanne M. (March 2019). "Smoking-induced Iron Dysregulation in the Lung". Free Radical Biology & Medicine. 133: 238–247. doi:10.1016/j.freeradbiomed.2018.07.024. ISSN 0891-5849. PMC 6355389. PMID 30075191.
- Boes, Katie M.; Durham, Amy C. (2017-01-01), Zachary, James F. (ed.), "Chapter 13 - Bone Marrow, Blood Cells, and the Lymphoid/Lymphatic System1", Pathologic Basis of Veterinary Disease (Sixth Edition), Mosby, pp. 724–804.e2, doi:10.1016/b978-0-323-35775-3.00013-8, ISBN 978-0-323-35775-3, PMC 7158316
- Greaves, Peter (2012-01-01), Greaves, Peter (ed.), "Chapter 6 - Respiratory Tract", Histopathology of Preclinical Toxicity Studies (Fourth Edition), Boston: Academic Press, pp. 207–261, doi:10.1016/b978-0-444-53856-7.00006-3, ISBN 978-0-444-53856-7, PMC 7151878
- Fukihara, Jun; Taniguchi, Hiroyuki; Ando, Masahiko; Kondoh, Yasuhiro; Kimura, Tomoki; Kataoka, Kensuke; Furukawa, Taiki; Johkoh, Takeshi; Fukuoka, Junya; Sakamoto, Koji; Hasegawa, Yoshinori (2017-02-06). "Hemosiderin-laden macrophages are an independent factor correlated with pulmonary vascular resistance in idiopathic pulmonary fibrosis: a case control study". BMC Pulmonary Medicine. 17 (1): 30. doi:10.1186/s12890-017-0376-8. ISSN 1471-2466. PMC 5294720. PMID 28166761.
- Strzelak, Agnieszka; Ratajczak, Aleksandra; Adamiec, Aleksander; Feleszko, Wojciech (May 2018). "Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review". International Journal of Environmental Research and Public Health. 15 (5): 1033. doi:10.3390/ijerph15051033. ISSN 1661-7827. PMC 5982072. PMID 29883409.
- Pankow, W.; Neumann, K.; Rüschoff, J.; von Wichert, P. (1995). "Human alveolar macrophages: comparison of cell size, autofluorescence, and HLA-DR antigen expression in smokers and nonsmokers". Cancer Detection and Prevention. 19 (3): 268–273. ISSN 0361-090X. PMID 7750115.
- Zmijewski, Jaroslaw W.; Pittet, Jean-Francois (October 2020). "Human Leukocyte Antigen-DR Deficiency and Immunosuppression-Related End-Organ Failure in SARS-CoV2 Infection". Anesthesia & Analgesia. 131 (4): 989–992. doi:10.1213/ANE.0000000000005140. ISSN 0003-2999. PMC 7386673. PMID 32925313.
- Jiang, Chen; Chen, Qiong; Xie, Mingxuan (2020-07-14). "Smoking increases the risk of infectious diseases: A narrative review". Tobacco Induced Diseases. 18: 60. doi:10.18332/tid/123845. ISSN 2070-7266. PMC 7398598. PMID 32765200.
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health (2014). The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Reports of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention (US). PMID 24455788.
- Thomson, N.C. (2004-11-01). "Asthma and cigarette smoking". European Respiratory Journal. 24 (5): 822–833. doi:10.1183/09031936.04.00039004. ISSN 0903-1936. PMID 15516679. S2CID 2941659.
- Zuo, Li; He, Feng; Sergakis, Georgianna G.; Koozehchian, Majid S.; Stimpfl, Julia N.; Rong, Yi; Diaz, Philip T.; Best, Thomas M. (2014-08-01). "Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments". American Journal of Physiology. Lung Cellular and Molecular Physiology. 307 (3): L205–L218. doi:10.1152/ajplung.00330.2013. ISSN 1040-0605. PMID 24879054.
- Walser, Tonya; Cui, Xiaoyan; Yanagawa, Jane; Lee, Jay M.; Heinrich, Eileen; Lee, Gina; Sharma, Sherven; Dubinett, Steven M. (2008-12-01). "Smoking and Lung Cancer". Proceedings of the American Thoracic Society. 5 (8): 811–815. doi:10.1513/pats.200809-100TH. ISSN 1546-3222. PMC 4080902. PMID 19017734.
- "Quit smoking - Better Heath". nhs.uk. 2020-11-03. Retrieved 2022-04-17.
- Feldman, Charles; Anderson, Ronald (2013-09-01). "Cigarette smoking and mechanisms of susceptibility to infections of the respiratory tract and other organ systems". Journal of Infection. 67 (3): 169–184. doi:10.1016/j.jinf.2013.05.004. ISSN 0163-4453. PMID 23707875.
- "Will my body ever get back to normal after I quit smoking? | Quit Smoking". Sharecare. Retrieved 2022-04-17.
- CDCTobaccoFree (2021-06-03). "Benefits of Quitting". Centers for Disease Control and Prevention. Retrieved 2022-04-17.
- Doll, Richard; Peto, Richard; Boreham, Jillian; Sutherland, Isabelle (2004-06-26). "Mortality in relation to smoking: 50 years' observations on male British doctors". BMJ: British Medical Journal. 328 (7455): 1519. doi:10.1136/bmj.38142.554479.AE. ISSN 0959-8138. PMC 437139. PMID 15213107.
- CDCTobaccoFree (2022-03-16). "About the Campaign". Centers for Disease Control and Prevention. Retrieved 2022-04-19.
- Gierisch, Jennifer M.; Bastian, Lori A.; Calhoun, Patrick S.; McDuffie, Jennifer R.; John W Williams, Jr (November 2010). FUTURE RESEARCH. Department of Veterans Affairs (US).
- "Challenges When Quitting | Smokefree". smokefree.gov. Retrieved 2022-04-18.