Somatic hypermutation
Somatic hypermutation (or SHM) is a cellular mechanism by which the immune system adapts to the new foreign elements that confront it (e.g. microbes), as seen during class switching. A major component of the process of affinity maturation, SHM diversifies B cell receptors used to recognize foreign elements (antigens) and allows the immune system to adapt its response to new threats during the lifetime of an organism.[1] Somatic hypermutation involves a programmed process of mutation affecting the variable regions of immunoglobulin genes. Unlike germline mutation, SHM affects only an organism's individual immune cells, and the mutations are not transmitted to the organism's offspring.[2] Because this mechanism is merely selective and not precisely targeted, somatic hypermutation has been strongly implicated in the development of B-cell lymphomas[3] and many other cancers.[4][5]
Targeting
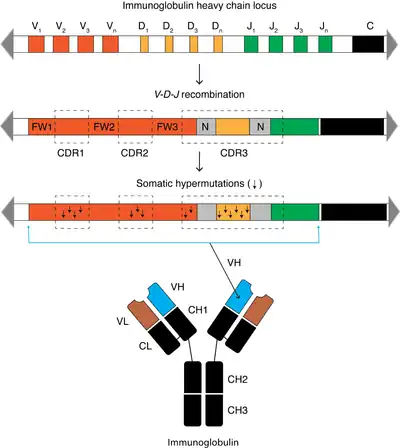
When a B cell recognizes an antigen, it is stimulated to divide (or proliferate). During proliferation, the B-cell receptor locus undergoes an extremely high rate of somatic mutation that is at least 105–106 fold greater than the normal rate of mutation across the genome.[2] Variation is mainly in the form of single-base substitutions, with insertions and deletions being less common. These mutations occur mostly at "hotspots" in the DNA, which are concentrated in hypervariable regions. These regions correspond to the complementarity-determining regions; the sites involved in antigen recognition on the immunoglobulin.[6] The "hotspots" of somatic hypermutation vary depending on the base that is being mutated. RGYW for a G, WRCY for a C, WA for an A and TW for a T.[7][8] The overall result of the hypermutation process is achieved by a balance between error-prone and high fidelity repair.[9] This directed hypermutation allows for the selection of B cells that express immunoglobulin receptors possessing an enhanced ability to recognize and bind a specific foreign antigen.[1]
Mechanisms
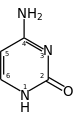
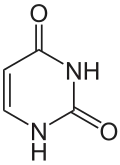
The mechanism of SHM involves deamination of cytosine to uracil in DNA by the enzyme activation-induced cytidine deaminase, or AID.[10][11] A cytosine:guanine pair is thus directly mutated to a uracil:guanine mismatch. Uracil residues are not normally found in DNA, therefore, to maintain the integrity of the genome, most of these mutations must be repaired by high-fidelity Base excision repair enzymes. The uracil bases are removed by the repair enzyme, uracil-DNA glycosylase.[11] Error-prone DNA polymerases are then recruited to fill in the gap and create mutations.[10][12]
The synthesis of this new DNA involves error-prone DNA polymerases, which often introduce mutations at the position of the deaminated cytosine itself or neighboring base pairs. During B cell division the immunoglobulin variable region DNA is transcribed and translated. The introduction of mutations in the rapidly proliferating population of B cells ultimately culminates in the production of thousands of B cells, possessing slightly different receptors and varying specificity for the antigen, from which the B cell with highest affinities for the antigen can be selected. The B cells with the greatest affinity will then be selected to differentiate into plasma cells producing antibody and long-lived memory B cells contributing to enhanced immune responses upon reinfection.[2]
The hypermutation process also utilizes cells that auto-select against the 'signature' of an organism's own cells. It is hypothesized that failures of this auto-selection process may also lead to the development of an auto-immune response.[13]
Models
Developments on the viability of the two main competing molecular models on the mechanism of somatic hypermutation (SHM) since 1987 have now reached a resolution, particular molecular data published since 2000. Much of this early phase data has been reviewed by Teng and Papavasiliou[10] and additionally outlined by Di Noia and Maul,[14][15] and the SHM field data reviewed in Steele[16][17] and additionally outlined in these papers.[4][5][17][18][19][20][21]
DNA deamination model
This can be labelled the DNA-based model. It is enzymatically focused solely on DNA substrates. The modern form, outlined in previous sections is the Neuberger "DNA deamination model" based on activation-induced cytidine deaminase (AID) and short-patch error-prone DNA repair by DNA polymerase-eta operating around AID C-to-U lesions[10][14][15] This model only partially explains the origins of the full spectrum of somatic mutations at A:T and G:C base pairs observed in SHM in B lymphocytes in vivo during an antigen-driven immune response. It also does not logically explain how strand biased mutations may be generated. A key feature is its critical dependence on the gap-filling error prone DNA repair synthesis properties of DNA polymerase-eta targeting A:T base pairs at AID-mediated C-to-U lesions or ssDNA nicks.[22][23][24] This error-prone DNA polymerase is the only known error-prone polymerase involved in SHM in vivo.[24] What is often ignored in these studies is that this Y family DNA polymerase enzyme is also an efficient reverse transcriptase as demonstrated in in vitro assays.[20]
Reverse transcriptase model
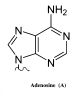
The more controversial competing mechanism is an RNA/RT-based mechanism (reverse transcriptase model of SHM) which attempts to explain the production of the full spectrum of strand-biased mutations at A:T and G:C base pairs whereby mutations of A are observed to exceed mutations of T (A>>>T) and mutations of G are observed to exceed mutations of C (G>>>C). This involves error-prone cDNA synthesis via an RNA-dependent DNA polymerase copying the base modified Ig pre-mRNA template and integrating the now error-filled cDNA copy back into the normal chromosomal site. The errors in the Ig pre-mRNA are a combination of adenosine-to-inosine (A-to-I) RNA editing[18][19] and RNA polymerase II transcription elongation complex copying uracil and abasic sites (arising as AID-mediated lesions) into the nascent pre-mRNA using the transcribed (TS) DNA as the copying template strand.[21] The modern form of this mechanism thus critically depends on AID C-to-U DNA lesions and long tract error-prone cDNA synthesis of the transcribed strand by DNA polymerase-eta acting as a reverse transcriptase.[16]
The evidence for and against each mechanism is critically evaluated in Steele[16] showing that all the molecular data on SHM published since 1980 supports directly or indirectly this RNA/RT-based mechanism. Recently Zheng et al.[25] have supplied critical independent validation by showing that Adenosine Deaminase enzymes acting on RNA (ADARs) can A-to-I edit both the RNA and DNA moieties of RNA:DNA hybrids in biochemical assays in vitro. RNA:DNA hybrids of about 11 nucleotides in length are transient structures formed at transcription bubbles in vivo during RNA polymerase II elongation.
An analysis of the implications of the Zheng et al. data has been published. [Steele, E.J.; Lindley, R.A. (2017). “ ADAR deaminase A-to-I editing of DNA and RNA moieties of RNA:DNA hybrids has implications for the mechanism of Ig somatic hypermutation.” DNA Repair. 55 (7): 1-6. doi:10.1016/j.dnarep.2017.04.004. PMID 28482199.] The Zheng et al.[25] data strongly imply that the RNA moiety would need to be first A-to-I RNA edited then reverse transcribed and integrated to generate the strong A>>>T strand biased mutation signatures at A:T base pairs observed in all SHM and cancer hypermutation data sets.[4][5][16][21] Editing (A-to-I) of the DNA moiety at RNA:DNA hybrids in vivo cannot explain the A>>T strand bias because such direct DNA modifications would result in T>>>A strand bias which is not observed in any SHM or cancer data set in vivo.[4][5][16][21] In this regard Robyn Lindley has also recently discovered that the Ig-SHM-like strand-biased mutations in cancer genome protein-coding genes are also in "codon-context". Lindley has termed this process targeted somatic mutation (TSM) to highlight that somatic mutations are far more targeted than previously thought in somatic tissues associated with disease.[26][27] The TSM process implies an "in-frame DNA reader" whereby DNA and RNA deaminases at transcribed regions are guided in their mutagenic action, by the codon reading frame of the DNA.[26][27]
The observation that DNA polymerase eta is an efficient reverse transcriptase Franklin, A.; Milburn, P. J.; Blanden, R.V.; Steele, E. J. (2004). "Human DNA polymerase-eta an A-T mutator in somatic hypermutation of rearranged immunoglobulin genes, is a reverse transcriptase". Immunol. Cell Biol. 82 (2): 219–225. doi:10.1046/j.0818-9641.2004.01221.x. PMID 15061777. S2CID 24370183. has been independently confirmed.[29] Su, Y.; M. Egli, M,;Guengerich. F.P (2017) “Human DNA polymerase η accommodates RNA for strand extension.” J. Biol. Chem. 292 (44) :18044–18051. DOI: 10.1074/jbc.M117.809723, PMID: 28972162 [30] Su, Y.; Ghodke, P.P.; Egli, M.; Li, L.; Wang, Y.; F.P. Guengerich, F.P. (2019) “ Human DNA polymerase η has reverse transcriptase activity in cellular environments.” J. Biol. Chem. 294 (15) : 6073–6081. DOI: 10.1074/jbc.RA119.007925 PMID: 30842261 Further the DNA repair enzyme DNA polymerase theta has also been shown to be an efficient cellular reverse transcriptase. [31 Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick.T. et. al. (2021) “ Polθ reverse transcribes RNA and promotes RNA-templated DNA repair.” Sci. Adv. 7, eabf1771. DOI: 10.1126/sciadv.abf1771 PMID: 34117057. Both these important observations have been reviewed in the context of the reverse transcriptase model of somatic hypermutation.[Franklin, A.; Steele, E.J. (2022) “ RNA-directed DNA repair and antibody somatic hypermutation.”. Trends Genet. 38 (5) 426-436. DOI: 10.1016/j.tig.2021.10.005 PMID: 34740453]
See also
References
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. (2005). Immunobiology (6th ed.). Garland Science. ISBN 978-0-8153-4101-7.
- Oprea, M. (1999) Antibody Repertoires and Pathogen Recognition: Archived 2008-09-06 at the Wayback Machine The Role of Germline Diversity and Somatic Hypermutation (Thesis) University of Leeds.
- Odegard V.H.; Schatz D.G. (2006). "Targeting of somatic hypermutation". Nat. Rev. Immunol. 6 (8): 573–583. doi:10.1038/nri1896. PMID 16868548. S2CID 6477436.
- Steele, E.J.; Lindley, R.A. (2010). "Somatic mutation patterns in non-lymphoid cancers resemble the strand biased somatic hypermutation spectra of antibody genes" (PDF). DNA Repair. 9 (6): 600–603. doi:10.1016/j.dnarep.2010.03.007. PMID 20418189.
- Lindley, R.A.; Steele, E.J. (2013). "Critical analysis of strand-biased somatic mutation signatures in TP53 versus Ig genes, in genome -wide data and the etiology of cancer". ISRN Genomics. 2013 Article ID 921418: 18 pages.
- Li, Z.; Wool, C.J.; Iglesias-Ussel; M.D., Ronai, D.; Scharff, M.D. (2004). "The generation of antibody diversity through somatic hypermutation and class switch recombination". Genes & Development. 18 (1): 1–11. doi:10.1101/gad.1161904. PMID 14724175.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Dunn-Walters, DK; Dogan, A; Boursier, L; MacDonald, CM; Spencer, J (1998). "Base-specific sequences that bias somatic hypermutation deduced by analysis of out of frame genes". J. Immunol. 160: 2360–64. doi:10.4049/jimmunol.160.5.2360. S2CID 23647692.
- Spencer, J; Dunn-Walters, DK (2005). "Hypermutation at A-T base pairs: The A nucleotide replacement spectrum is affected by adjacent nucleotides and there is no reverse complementarity of sequences around A and T nucleotides". J. Immunol. 175 (8): 5170–77. doi:10.4049/jimmunol.175.8.5170. PMID 16210621.
- Liu, M.; Schatz, D.G. (2009). "Balancing AID and DNA repair during somatic hypermutation". Trends in Immunology. 30 (4): 173–181. doi:10.1016/j.it.2009.01.007. PMID 19303358.
- Teng, G.; Papavasiliou, F.N. (2007). "Immunoglobulin Somatic Hypermutation". Annu. Rev. Genet. 41: 107–120. doi:10.1146/annurev.genet.41.110306.130340. PMID 17576170.
- Larson, E.D.; Maizels, N. (2004). "Transcription-coupled mutagenesis by the DNA deaminase AID". Genome Biol. 5 (3): 211. doi:10.1186/gb-2004-5-3-211. PMC 395756. PMID 15003109.
- Bachl, J.; Ertongur, I.; Jungnickel, B. (2006). "Involvement of Rad18 in somatic hypermutation". Proc. Natl. Acad. Sci. USA. 103 (32): 12081–86. Bibcode:2006PNAS..10312081B. doi:10.1073/pnas.0605146103. PMC 1567700. PMID 16873544.
- Metzger, T.C. (2011). "Control of Central and Peripheral Tolerance by Aire". Immunological Reviews. 241 (1): 89–103. doi:10.1111/j.1600-065X.2011.01008.x. PMC 3093413. PMID 21488892.
- Di Noia, J. M.; Neuberger, M. S. (2007). "Molecular mechanisms of somatic hypermutation". Annu. Rev. Biochem. 76: 1–22. doi:10.1146/annurev.biochem.76.061705.090740. PMID 17328676.
- Maul, R. W.; Gearhart, P. J. (2010). AID and Somatic Hypermutation. Advances in Immunology. Vol. 105. pp. 159–191. doi:10.1016/S0065-2776(10)05006-6. ISBN 9780123813022. PMC 2954419. PMID 20510733.
- Steele, E.J. (2016). "Somatic hypermutation in immunity and cancer: Critical analysis of strand-biased and codon-context mutation signatures". DNA Repair. 45: 1–24. doi:10.1016/j.dnarep.2016.07.001. PMID 27449479.
- Steele, E.J.; Pollard, J.W. (1987). "Hypothesis : Somatic Hypermutation by gene conversion via the error prone DNA-to-RNA-to-DNA information loop". Mol. Immunol. 24 (6): 667–673. doi:10.1016/j.dnarep.2016.07.001. PMID 2443841.
- Steele, E.J.; Lindley, R.A.; Wen, J; Weiler, G.F. (2006). "Computational analyses show A-to-G mutations correlate with nascent mRNA hairpins at somatic hypermutation hotspots". DNA Repair. 5 (11): 1346–1363. doi:10.1016/j.dnarep.2006.06.002. PMID 16884961.
- Steele, E.J.; Franklin, A; Blanden, R.V. (2004). "Genesis of the strand biased signature in somatic hypermutation of rearranged immunoglobulin variable genes". Immunol. Cell Biol. 82 (2): 208–218. doi:10.1046/j.0818-9641.2004.01224.x. PMID 15061776. S2CID 23764779.
- Franklin, A.; Milburn, P. J.; Blanden, R.V.; Steele, E. J. (2004). "Human DNA polymerase-eta an A-T mutator in somatic hypermutation of rearranged immunoglobulin genes, is a reverse transcriptase". Immunol. Cell Biol. 82 (2): 219–225. doi:10.1046/j.0818-9641.2004.01221.x. PMID 15061777. S2CID 24370183.
- Steele, E.J. (2009). "Mechanism of somatic hypermutation: Critical analysis of strand biased mutation signatures at A:T and G:C base pairs". Mol. Immunol. 46 (3): 305–320. doi:10.1016/j.molimm.2008.10.021. PMID 19062097.
- Zeng, X; Winter, D.B.; Kasmer, C; Kraemer, K.H.; Lehmann, A.R.; Gearhart, P.J. (2001). "DNA polymerase-eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes". Nat. Immunol. 2 (6): 537–541. doi:10.1038/88740. PMID 11376341. S2CID 6213513.
- Wilson, T.M.; Vaisman, A; Martomo, S.A.; Sullivan, P; Lan, L.; Hanaoka, F.; Yasui, A.; Woodgate, R.; Gearhart, P.J. (2005). "MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes". J. Exp. Med. 201 (4): 637–645. doi:10.1084/jem.20042066. PMC 2213055. PMID 15710654.
- Delbos, F; Aoufouchi, S; Faili, A; Weill, J-C; Reynaud, C-A (2007). "DNA polymerase-eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse". J. Exp. Med. 204 (2007): 17–23. doi:10.1084/jem.20062131. PMC 2118439. PMID 17190840.
- Zheng, Yuxuan; Lorenzo, Claire; Beal, Peter A. (27 January 2017). "DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA". Nucleic Acids Research. 45 (6): 3369–337. doi:10.1093/nar/gkx050. PMC 5389660. PMID 28132026.
- Lindley, R.A. (2013). "The importance of codon context for understanding the Ig-like somatic hypermutation strand-biased patterns in TP53 mutations in breast cancer". Cancer Genet. 206 (6): 222–226. doi:10.1016/j.cancergen.2013.05.016. PMID 23880211.
- Lindley, R.A.; Humbert, P; Larmer, C; Akmeemana, E.H.; Pendlebury, C.R.R. (2016). "Association between Targeted Somatic Mutation (TSM) signatures and HGS-OvCa progression". Cancer Med. 5 (9): 2629–2640. doi:10.1002/cam4.825. PMC 5055158. PMID 27485054.
20 Franklin, A.; Milburn, P. J.; Blanden, R.V.; Steele, E. J. (2004). "Human DNA polymerase-eta an A-T mutator in somatic hypermutation of rearranged immunoglobulin genes, is a reverse transcriptase". Immunol. Cell Biol. 82 (2): 219–225. doi:10.1046/j.0818-9641.2004.01221.x. PMID 15061777. S2CID 24370183.
External links
- Immunoglobulin+somatic+hypermutation at the U.S. National Library of Medicine Medical Subject Headings (MeSH)