Splice site mutation
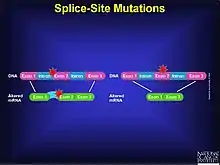
A splice site mutation is a genetic mutation that inserts, deletes or changes a number of nucleotides in the specific site at which splicing takes place during the processing of precursor messenger RNA into mature messenger RNA. Splice site consensus sequences that drive exon recognition are located at the very termini of introns.[1] The deletion of the splicing site results in one or more introns remaining in mature mRNA and may lead to the production of abnormal proteins. When a splice site mutation occurs, the mRNA transcript possesses information from these introns that normally should not be included. Introns are supposed to be removed, while the exons are expressed.
The mutation must occur at the specific site at which intron splicing occurs: within non-coding sites in a gene, directly next to the location of the exon. The mutation can be an insertion, deletion, frameshift, etc. The splicing process itself is controlled by the given sequences, known as splice-donor and splice-acceptor sequences, which surround each exon. Mutations in these sequences may lead to retention of large segments of intronic DNA by the mRNA, or to entire exons being spliced out of the mRNA. These changes could result in production of a nonfunctional protein.[2] An intron is separated from its exon by means of the splice site. Acceptor-site and donor-site relating to the splice sites signal to the spliceosome where the actual cut should be made. These donor sites, or recognition sites, are essential in the processing of mRNA. The average vertebrate gene consists of multiple small exons (average size, 137 nucleotides) separated by introns that are considerably larger.[1]
Background
In 1993, Richard J. Roberts and Phillip Allen Sharp received the Nobel Prize in Physiology or Medicine for their discovery of "split genes".[4] Using the model adenovirus in their research, they were able to discover splicing—the fact that pre-mRNA is processed into mRNA once introns were removed from the RNA segment. These two scientists discovered the existence of splice sites, thereby changing the face of genomics research. They also discovered that the splicing of the messenger RNA can occur in different ways, opening up the possibility for a mutation to occur.
Technology
Today, many different types of technologies exist in which splice sites can be located and analyzed for more information. The Human Splicing Finder is an online database stemming from the Human Genome Project data. The genome database identifies thousands of mutations related to medical and health fields, as well as providing critical research information regarding splice site mutations. The tool specifically searches for pre-mRNA splicing errors, the calculation of potential splice sites using complex algorithms, and correlation with several other online genomic databases, such as the Ensembl genome browser.[5]
Role in Disease
Due to the sensitive location of splice sites, mutations in the acceptor or donor areas of splice sites can become detrimental to a human individual. In fact, many different types of diseases stem from anomalies within the splice sites.
Cancer
A study researching the role of splice site mutations in cancer supported that a splice site mutation was common in a set of women who were positive for breast and ovarian cancer. These women had the same mutation, according to the findings. An intronic single base-pair substitution destroys an acceptor site, thus activating a cryptic splice site, leading to a 59 base-pair insertion and chain termination. The four families with both breast and ovarian cancer had chain termination mutations in the N-terminal half of the protein.[6] The mutation in this research example was located within the splice-site.
Dementia
According to a research study conducted Hutton, M et al, a missense mutation occurring on the 5' region of the RNA associated with the tau protein was found to be correlated with inherited dementia (known as FTDP-17). The splice-site mutations all destabilize a potential stem–loop structure which is most likely involved in regulating the alternative splicing of exon10 in chromosome 17. Consequently, more usage occurs on the 5' splice site and an increased proportion of tau transcripts that include exon 10 are created. Such drastic increase in mRNA will increase the proportion of Tau containing four microtubule-binding repeats, which is consistent with the neuropathology described in several families with FTDP-17, a type inherited dementia.[7]
Epilepsy
Some types of epilepsy may be brought on due to a splice site mutation. In addition to a mutation in a stop codon, a splice site mutation on the 3' strand was found in a gene coding for cystatin B in Progressive Myoclonus Epilepsy[8] patients. This combination of mutations was not found in unaffected individuals. By comparing sequences with and without the splice site mutation, investigators were able to determine that a G-to-C nucleotide transversion occurs at the last position of the first intron. This transversion occurs in the region that codes for the cystatin B gene. Individuals suffering from Progressive Myoclonus Epilepsy possess a mutated form of this gene, which results in decreased output of mature mRNA, and subsequently decreases in protein expression.
A study has also shown that a type of Childhood Absence Epilepsy (CAE) causing febrile seizures may be linked to a splice site mutation in the sixth intron of the GABRG2 gene. This splice site mutation was found to cause a nonfunctional GABRG2 subunit in affected individuals.[9] According to this study, a point mutation was the culprit for the splice-donor site mutation, which occurred in intron 6. A nonfunctional protein product is produced, leading to the also nonfunctional subunit.
Hematological Disorders
Several genetic diseases may be the result of splice site mutations. For example, mutations that cause the incorrect splicing of β-globin mRNA are responsible of some cases of β-thalassemia. Another Example is TTP (thrombotic thrombocytopenic purpura). TTP is caused by deficiency of ADAMTS-13. A splice site mutation of ADAMTS-13 gene can therefore cause TTP. It is estimated that 15% of all point mutations causing human genetic diseases occur within a splice site.[10]
Parathyroid Deficiency
When a splice site mutation occurs in intron 2 of the gene that produces the parathyroid hormone, a parathyroid deficiency can prevail. In one particular study, a G to C substitution in the splice site of intron 2 produces a skipping effect in the messenger RNA transcript. The exon that is skipped possesses the initiation start codon to produce parathyroid hormone.[11] Such failure in initiation causes the deficiency.
Analysis
Using the model organism Drosophila melanogaster, data has been compiled regarding the genomic information and sequencing of this organism. A prediction model exists in which a researcher can upload his or her genomic information and use a splice site prediction database to gather information about where the splice sites could be located. The Berkeley Drosophila Project can be used to incorporate this research, as well as annotate high quality euchromatic data. The splice site predictor can be a great tool for researchers studying human disease in this model organism.
Splice site mutations can be analyzed using information theory.[12]
References
- Berget, Susan (1995). "Exon Recognition in Vertebrate Splicing". The Journal of Biological Chemistry. 270 (6): 2411–2414. doi:10.1074/jbc.270.6.2411. PMID 7852296.
- Understanding Cancer Genomics: Splice Site Mutations. National Cancer Institute.
- Understanding Cancer Genomics. National Cancer Institute.
- "Physiology or Medicine 1993 - Press Release". www.nobelprize.org. Retrieved 2017-10-07.
- "The Human Splicing Finder".
- Friedman, Lori (1994). "Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families". Nature Genetics. 8 (4): 399–404. doi:10.1038/ng1294-399. PMID 7894493. S2CID 2863113.
- Hutton, Mike; Lendon, Corinne; Rizzu, Patrizia; Baker, Matt (18 June 1998). "Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17". Nature. 393 (6686): 702–705. Bibcode:1998Natur.393..702H. doi:10.1038/31508. PMID 9641683. S2CID 205001265.
- Pennacchio, Len A.; Lehesjoki, Anna-Elina; Stone, Nancy E.; Wilour, Virginia L. (Mar 22, 1996). "Mutations in the Gene Encoding Cystatin B in Progressive Myoclonus Epilepsy (EPM1)". Science. 271 (5256): 1731–1734. Bibcode:1996Sci...271.1731P. doi:10.1126/science.271.5256.1731. JSTOR 2890839. PMID 8596935. S2CID 84361089.
- Kananura, Colette; Haug, Karsten; Sander, Thomas; Runge, Uwe; Gu, Wenli; Hallmann, Kerstin; Rebstock, Johannes; Heils, Armin; Steinlein, Ortrud (July 2002). "A Splice-Site Mutation in GABRG2 Associated With Childhood Absence Epilepsy and Febrile Convulsions". Arch Neurol. 59 (7): 1137–1141. doi:10.1001/archneur.59.7.1137. PMID 12117362.
- Carvalho, Gisah A; Weiss, Roy E; Refetoff, Samuel (June 22, 1998). "Complete Thyroxine-Binding Globulin (TBG) Deficiency Produced by a Mutation in Acceptor Splice Site Causing Frameshift and Early Termination of Translation (TBG-Kankakee)". The Journal of Clinical Endocrinology and Metabolism. 83 (10): 3604–3608. doi:10.1210/jcem.83.10.5208. PMID 9768672.
- Parkinson, David B.; Thakker, Rajesh V. (1992). "A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism". Nature Genetics. 1 (2): 149–152. doi:10.1038/ng0592-149. PMID 1302009. S2CID 24032313.
- Rogan, PK; Faux, BM; Schneider, TD (1998). "Information analysis of human splice site mutations". Hum. Mutat. 12 (3): 153–71. doi:10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. PMID 9711873.