Staurosporine
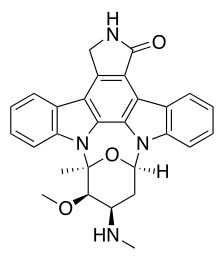
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus.[1] It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.[2]
 | |
 | |
| Clinical data | |
|---|---|
| ATC code |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.109.946 |
| Chemical and physical data | |
| Formula | C28H26N4O3 |
| Molar mass | 466.541 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive.[3] The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment.
Biological activities
The main biological activity of staurosporine is the inhibition of protein kinases through the prevention of ATP binding to the kinase. This is achieved through the stronger affinity of staurosporine to the ATP-binding site on the kinase. Staurosporine is a prototypical ATP-competitive kinase inhibitor in that it binds to many kinases with high affinity, though with little selectivity.[4] Structural analysis of kinase pockets demonstrated that main chain atoms which are conserved in their relative positions to staurosporine contributes to staurosporine promiscuity.[5] This lack of specificity has precluded its clinical use, but has made it a valuable research tool. In research, staurosporine is used to induce apoptosis. The mechanism of how it mediates this is not well understood. It has been found that one way in which staurosporine induces apoptosis is by activating caspase-3.[6] At lower concentration, depending on the cell type, staurosporine induces specific cell cycle effects arresting cells either in G1 or in G2 phase of the cell cycle.[7]
Chemistry family
Staurosporine is an indolocarbazole. It belongs to the most frequently isolated group of indolocarbazoles: Indolo(2,3-a)carbazoles. Of these, Staurosporine falls within the most common subgroup, called Indolo(2,3-a)pyrrole(3,4-c)carbazoles. These fall into two classes - halogenated (chlorinated) and non-halogenated. Halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond, while non-halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have both indole nitrogens glycosylated, and a fully reduced C-7 carbon. Staurosporine is in the non-halogenated class.[8]
Staurosporine is the precursor of the novel protein kinase inhibitor midostaurin (PKC412).[9][10] Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
pyrrole(3%252C4-c)carbazol.svg.png.webp)
Biosynthesis
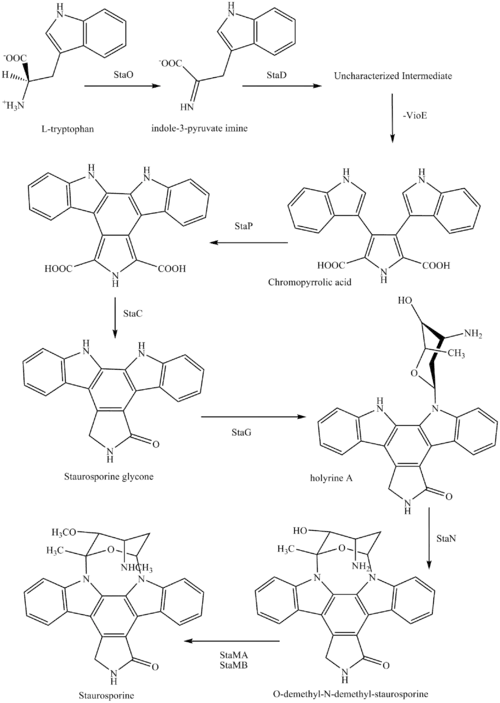
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imine by enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450 enzyme to form an aromatic ring system occurs.[8]
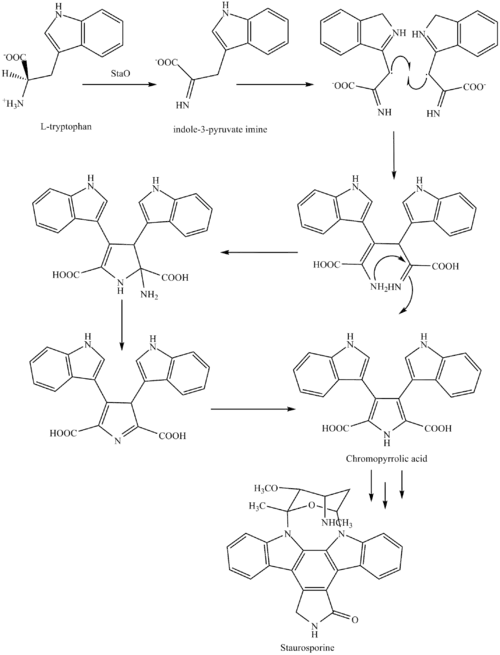
This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4'amine by StaMA and N-methylation of the 3'-hydroxy by StaMB leads to the formation of staurosporine.[8]
Research in preclinical use
When encapsulated in liposome nanoparticle, staurosporine is shown to suppress tumors in vivo in a mouse model without the toxic side effects which have prohibited its use as an anti-cancer drug with high apoptotic activity. Researchers in UC San Diego Moores Cancer Center develop a platform technology of high drug-loading efficiency by manipulating the pH environment of the cells. When injected into the mouse glioblastoma model, staurosporine is found to accumulate primarily in the tumor via fluorescence confirmation, and the mice did not suffer weight loss compared to the control mice administered with the free compound, an indicator of reduced toxicity.[11][12]
List of compounds closely related to Staurosporine
References
- Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchya H, et al. (April 1977). "A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization". The Journal of Antibiotics. 30 (4): 275–282. doi:10.7164/antibiotics.30.275. PMID 863788.
- Funato N, Takayanagi H, Konda Y, Toda Y, Harigaya Y, Omura S (1994). "Absolute configuration of staurosporine by X-ray analysis". Tetrahedron Lett. 35 (8): 1251–1254. doi:10.1016/0040-4039(94)88036-0.
- Rüegg UT, Burgess GM (June 1989). "Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases". Trends in Pharmacological Sciences. 10 (6): 218–20. doi:10.1016/0165-6147(89)90263-0. PMID 2672462.
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. (January 2008). "A quantitative analysis of kinase inhibitor selectivity". Nature Biotechnology. 26 (1): 127–132. doi:10.1038/nbt1358. PMID 18183025. S2CID 205273598.
- Tanramluk D, Schreyer A, Pitt WR, Blundell TL (July 2009). "On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine". Chemical Biology & Drug Design. 74 (1): 16–24. doi:10.1111/j.1747-0285.2009.00832.x. PMC 2737611. PMID 19519740.
- Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, et al. (October 2000). "Molecular mechanism of staurosporine-induced apoptosis in osteoblasts". Pharmacological Research. 42 (4): 373–381. doi:10.1006/phrs.2000.0700. PMID 10987998.
- Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z (January 1992). "Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes". Cancer Research. 52 (2): 470–473. PMID 1728418.
- Ryan KS (2008). "Structural studies of rebeccamycin, staurosporine, and violacein biosynthetic enzymes" (PDF). Ph.D. Thesis. Massachusetts Institute of Technology. Archived from the original (PDF) on 2012-03-14.
- Midostaurin product page, Fermentek
- Wang Y, Yin OQ, Graf P, Kisicki JC, Schran H (June 2008). "Dose- and time-dependent pharmacokinetics of midostaurin in patients with diabetes mellitus". Journal of Clinical Pharmacology. 48 (6): 763–775. doi:10.1177/0091270008318006. PMID 18508951. S2CID 26657407.
- News Release (21 October 2013). "Study Identifies Safe Delivery System for Tricky Yet Highly Potent Anti-Cancer Compounds". UC San Diego Health System. Retrieved 27 October 2013.
- Mukthavaram R, Jiang P, Saklecha R, Simberg D, Bharati IS, Nomura N, et al. (2013). "High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile". International Journal of Nanomedicine. 8 (1): 3991–4006. doi:10.2147/IJN.S51949. PMC 3808212. PMID 24174874.