Asymmetric induction
Asymmetric induction (also enantioinduction) describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment.[1] Asymmetric induction is a key element in asymmetric synthesis.

Asymmetric induction was introduced by Hermann Emil Fischer based on his work on carbohydrates.[2] Several types of induction exist.
Internal asymmetric induction makes use of a chiral center bound to the reactive center through a covalent bond and remains so during the reaction. The starting material is often derived from chiral pool synthesis. In relayed asymmetric induction the chiral information is introduced in a separate step and removed again in a separate chemical reaction. Special synthons are called chiral auxiliaries. In external asymmetric induction chiral information is introduced in the transition state through a catalyst of chiral ligand. This method of asymmetric synthesis is economically most desirable.
Carbonyl 1,2 asymmetric induction
Several models exist to describe chiral induction at carbonyl carbons during nucleophilic additions. These models are based on a combination of steric and electronic considerations and are often in conflict with each other. Models have been devised by Cram (1952), Cornforth (1959), Felkin (1969) and others.
Cram's rule
The Cram's rule of asymmetric induction developed by Donald J. Cram in 1952[3] is an early concept relating to the prediction of stereochemistry in certain acyclic systems. In full the rule is:
In certain non-catalytic reactions that diastereomer will predominate, which could be formed by the approach of the entering group from the least hindered side when the rotational conformation of the C-C bond is such that the double bond is flanked by the two least bulky groups attached to the adjacent asymmetric center.
The rule indicates that the presence of an asymmetric center in a molecule induces the formation of an asymmetric center adjacent to it based on steric hindrance.
In his 1952 publication Cram presented a large number of reactions described in the literature for which the conformation of the reaction products could be explained based on this rule and he also described an elaborate experiment (scheme 1) making his case.
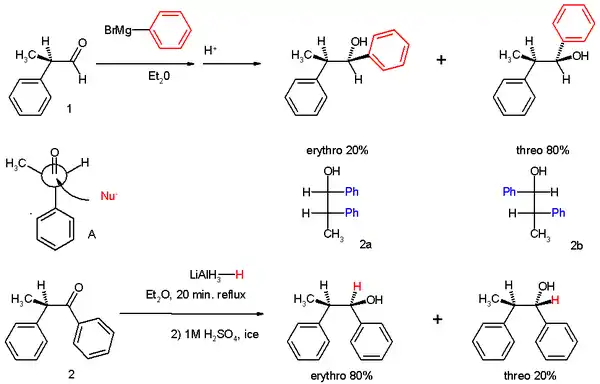
The experiments involved two reactions. In experiment one 2-phenylpropionaldehyde (1, racemic but (R)-enantiomer shown) was reacted with the Grignard reagent of bromobenzene to 1,2-diphenyl-1-propanol (2) as a mixture of diastereomers, predominantly the threo isomer (see for explanation the Fischer projection).
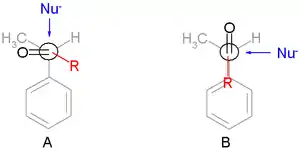
The preference for the formation of the threo isomer can be explained by the rule stated above by having the active nucleophile in this reaction attacking the carbonyl group from the least hindered side (see Newman projection A) when the carbonyl is positioned in a staggered formation with the methyl group and the hydrogen atom, which are the two smallest substituents creating a minimum of steric hindrance, in a gauche orientation and phenyl as the most bulky group in the anti conformation.
The second reaction is the organic reduction of 1,2-diphenyl-1-propanone 2 with lithium aluminium hydride, which results in the same reaction product as above but now with preference for the erythro isomer (2a). Now a hydride anion (H−) is the nucleophile attacking from the least hindered side (imagine hydrogen entering from the paper plane).
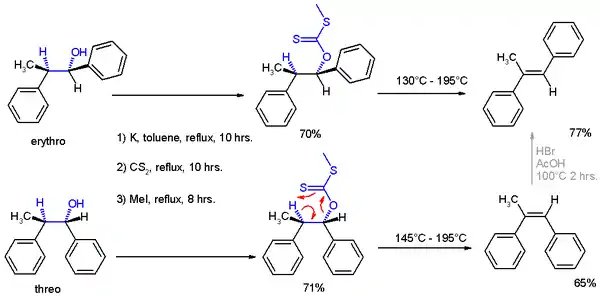
In the original 1952 publication, additional evidence was obtained for the structural assignment of the reaction products by applying them to a Chugaev elimination, wherein the threo isomer reacts to the cis isomer of -α-methyl-stilbene and the erythro isomer to the trans version.
Felkin model
The Felkin model (1968) named after Hugh Felkin also predicts the stereochemistry of nucleophilic addition reactions to carbonyl groups.[4] Felkin argued that the Cram model suffered a major drawback: an eclipsed conformation in the transition state between the carbonyl substituent (the hydrogen atom in aldehydes) and the largest α-carbonyl substituent. He demonstrated that by increasing the steric bulk of the carbonyl substituent from methyl to ethyl to isopropyl to isobutyl, the stereoselectivity also increased, which is not predicted by Cram's rule:
The Felkin rules are:
- The transition states are reactant-like.
- Torsional strain (Pitzer strain) involving partial bonds (in transition states) represents a substantial fraction of the strain between fully formed bonds, even when the degree of bonding is quite low. The conformation in the TS is staggered and not eclipsed with the substituent R skew with respect to two adjacent groups one of them the smallest in TS A.
- For comparison TS B is the Cram transition state.
- The main steric interactions involve those around R and the nucleophile but not the carbonyl oxygen atom.
- Attack of the nucleophile occurs according to the Dunitz angle (107 degrees), eclipsing the hydrogen, rather than perpendicular to the carbonyl.
- A polar effect or electronic effect stabilizes a transition state with maximum separation between the nucleophile and an electron-withdrawing group. For instance haloketones do not obey Cram's rule, and, in the example above, replacing the electron-withdrawing phenyl group by a cyclohexyl group reduces stereoselectivity considerably.
Felkin–Anh model
The Felkin–Anh model[5] is an extension of the Felkin model that incorporates improvements suggested by Nguyễn Trọng Anh and Odile Eisenstein to correct for two key weaknesses in Felkin's model. The first weakness addressed was the statement by Felkin of a strong polar effect in nucleophilic addition transition states, which leads to the complete inversion of stereochemistry by SN2 reactions, without offering justifications as to why this phenomenon was observed. Anh's solution was to offer the antiperiplanar effect as a consequence of asymmetric induction being controlled by both substituent and orbital effects.[6][7] In this effect, the best nucleophile acceptor σ* orbital is aligned parallel to both the π and π* orbitals of the carbonyl, which provide stabilization of the incoming anion.
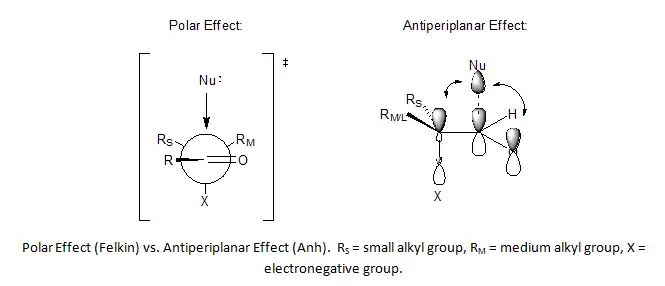
The second weakness in the Felkin Model was the assumption of substituent minimization around the carbonyl R, which cannot be applied to aldehydes.
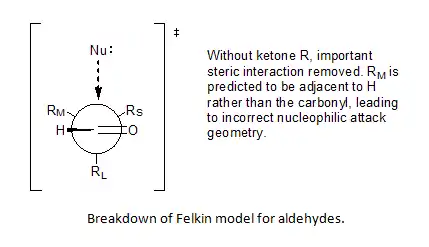
Incorporation of Bürgi–Dunitz angle[8][9] ideas allowed Anh to postulate a non-perpendicular attack by the nucleophile on the carbonyl center, anywhere from 95° to 105° relative to the oxygen-carbon double bond, favoring approach closer to the smaller substituent and thereby solve the problem of predictability for aldehydes.[6][10][11]
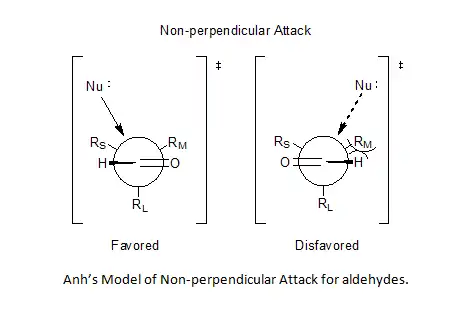
Anti–Felkin selectivity
Though the Cram and Felkin–Anh models differ in the conformers considered and other assumptions, they both attempt to explain the same basic phenomenon: the preferential addition of a nucleophile to the most sterically favored face of a carbonyl moiety. However, many examples exist of reactions that display stereoselectivity opposite of what is predicted by the basic tenets of the Cram and Felkin–Anh models. Although both of the models include attempts to explain these reversals, the products obtained are still referred to as "anti-Felkin" products. One of the most common examples of altered asymmetric induction selectivity requires an α-carbon substituted with a component with Lewis base character (i.e. O, N, S, P substituents). In this situation, if a Lewis acid such as Al-iPr2 or Zn2+ is introduced, a bidentate chelation effect can be observed. This locks the carbonyl and the Lewis base substituent in an eclipsed conformation, and the nucleophile will then attack from the side with the smallest free α-carbon substituent.[12] If the chelating R group is identified as the largest, this will result in an "anti-Felkin" product.
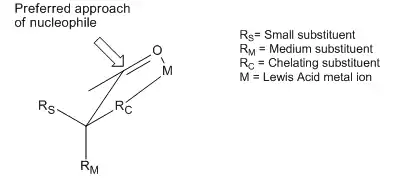
This stereoselective control was recognized and discussed in the first paper establishing the Cram model, causing Cram to assert that his model requires non-chelating conditions.[13] An example of chelation control of a reaction can be seen here, from a 1987 paper that was the first to directly observe such a "Cram-chelate" intermediate,[14] vindicating the model:
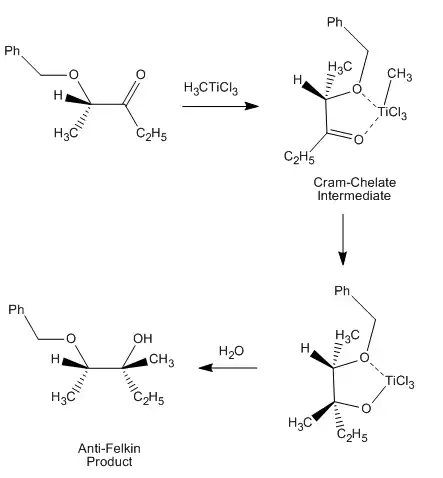
Here, the methyl titanium chloride forms a Cram-chelate. The methyl group then dissociates from titanium and attacks the carbonyl, leading to the anti-Felkin diastereomer.
A non-chelating electron-withdrawing substituent effect can also result in anti-Felkin selectivity. If a substituent on the α-carbon is sufficiently electron withdrawing, the nucleophile will add anti- relative to the electron withdrawing group, even if the substituent is not the largest of the 3 bonded to the α-carbon. Each model offers a slightly different explanation for this phenomenon. A polar effect was postulated by the Cornforth model[15] and the original Felkin model,[16] which placed the EWG substituent and incoming nucleophile anti- to each other in order to most effectively cancel the dipole moment of the transition structure.
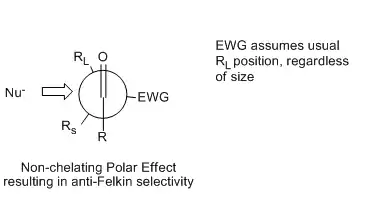
This Newman projection illustrates the Cornforth and Felkin transition state that places the EWG anti- to the incoming nucleophile, regardless of its steric bulk relative to RS and RL.
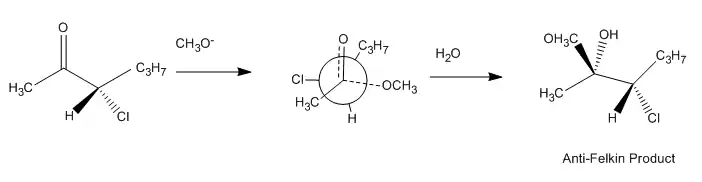
The improved Felkin–Anh model, as discussed above, makes a more sophisticated assessment of the polar effect by considering molecular orbital interactions in the stabilization of the preferred transition state. A typical reaction illustrating the potential anti-Felkin selectivity of this effect, along with its proposed transition structure, is pictured below:
Carbonyl 1,3 asymmetric induction
It has been observed that the stereoelectronic environment at the β-carbon of can also direct asymmetric induction. A number of predictive models have evolved over the years to define the stereoselectivity of such reactions.
Chelation model
According to Reetz, the Cram-chelate model for 1,2-inductions can be extended to predict the chelated complex of a β-alkoxy aldehyde and metal. The nucleophile is seen to attack from the less sterically hindered side and anti- to the substituent Rβ, leading to the anti-adduct as the major product.[17]
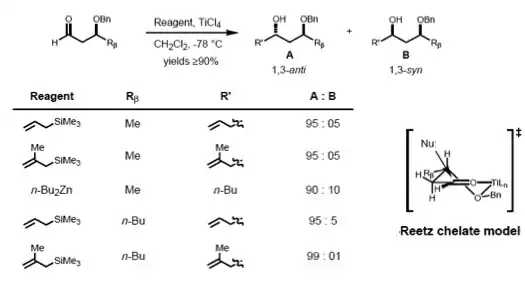
To make such chelates, the metal center must have at least two free coordination sites and the protecting ligands should form a bidentate complex with the Lewis acid.
Cram–Reetz model
Cram and Reetz demonstrated that 1,3-stereocontrol is possible if the reaction proceeds through an acyclic transition state. The reaction of β-alkoxy aldehyde with allyltrimethylsilane showed good selectivity for the anti-1,3-diol, which was explained by the Cram polar model. The polar benzyloxy group is oriented anti to the carbonyl to minimize dipole interactions and the nucleophile attacks anti- to the bulkier (RM) of the remaining two substituents.[18][19]
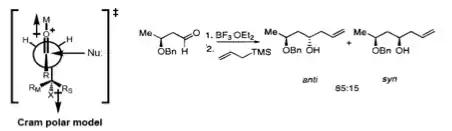
Evans model
More recently, Evans presented a different model for nonchelate 1,3-inductions. In the proposed transition state, the β-stereocenter is oriented anti- to the incoming nucleophile, as seen in the Felkin–Anh model. The polar X group at the β-stereocenter is placed anti- to the carbonyl to reduce dipole interactions, and Rβ is placed anti- to the aldehyde group to minimize the steric hindrance. Consequently, the 1,3-anti-diol would be predicted as the major product.[20]
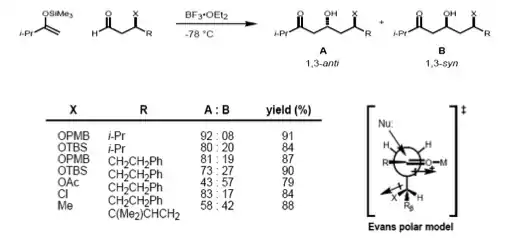
Carbonyl 1,2 and 1,3 asymmetric induction
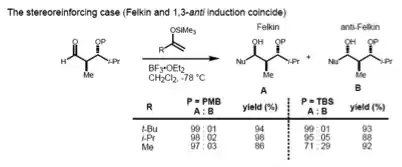
If the substrate has both an α- and β-stereocenter, the Felkin–Anh rule (1,2-induction) and the Evans model (1,3-induction) should considered at the same time. If these two stereocenters have an anti- relationship, both models predict the same diastereomer (the stereoreinforcing case).
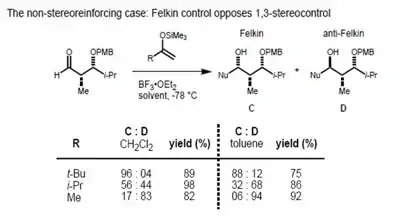
However, in the case of the syn-substrate, the Felkin–Anh and the Evans model predict different products (non-stereoreinforcing case). It has been found that the size of the incoming nucleophile determines the type of control exerted over the stereochemistry. In the case of a large nucleophile, the interaction of the α-stereocenter with the incoming nucleophile becomes dominant; therefore, the Felkin product is the major one. Smaller nucleophiles, on the other hand, result in 1,3 control determining the asymmetry.[21]
Acyclic alkenes asymmetric induction
Chiral acyclic alkenes also show diastereoselectivity upon reactions such as epoxidation and enolate alkylation. The substituents around the alkene can favour the approach of the electrophile from one or the other face of the molecule. This is the basis of the Houk's model, based on theoretical work by Kendall Houk, which predicts that the selectivity is stronger for cis than for trans double bonds.[22]
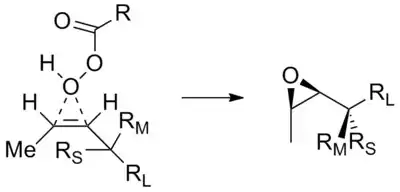
In the example shown, the cis alkene assumes the shown conformation to minimize steric clash between RS and the methyl group. The approach of the electrophile preferentially occurs from the same side of the medium group (RM) rather than the large group (RL), mainly producing the shown diastereoisomer. Since for a trans alkene the steric hindrance between RS and the H group is not as large as for the cis case, the selectivity is much lower.
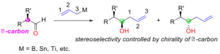
Substrate control: asymmetric induction by molecular framework in acyclic systems
Asymmetric induction by the molecular framework of an acyclic substrate is the idea that asymmetric steric and electronic properties of a molecule may determine the chirality of subsequent chemical reactions on that molecule. This principal is used to design chemical syntheses where one stereocentre is in place and additional stereocentres are required.
When considering how two functional groups or species react, the precise 3D configurations of the chemical entities involved will determine how they may approach one another. Any restrictions as to how these species may approach each other will determine the configuration of the product of the reaction. In the case of asymmetric induction, we are considering the effects of one asymmetric centre on a molecule on the reactivity of other functional groups on that molecule. The closer together these two sites are, the larger an influence is expected to be observed. A more holistic approach to evaluating these factors is by computational modelling,[23] however, simple qualitative factors may also be used to explain the predominant trends seen for some synthetic steps. The ease and accuracy of this qualitative approach means it is more commonly applied in synthesis and substrate design. Examples of appropriate molecular frameworks are alpha chiral aldehydes and the use of chiral auxiliaries.
Asymmetric induction at alpha-chiral aldehydes
Possible reactivity at aldehydes include nucleophilic attack and addition of allylmetals. The stereoselectivity of nucleophilic attack at alpha-chiral aldehydes may be described by the Felkin–Anh or polar Felkin Anh models and addition of achiral allylmetals may be described by Cram’s rule.
Felkin–Anh and polar Felkin–Anh model
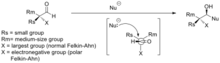
Selectivity in nucleophilic additions to chiral aldehydes is often explained by the Felkin–Anh model[24] (see figure). The nucleophile approaches the carbon of the carbonyl group at the Burgi-Dunitz angle.[25] At this trajectory, attack from the bottom face is disfavored due to steric bulk of the adjacent, large, functional group.
The polar Felkin–Anh model is applied in the scenario where X is an electronegative group. The polar Felkin–Anh model postulates that the observed stereochemistry arises due to hyperconjugative stabilization arising from the anti-periplanar interaction between the C-X antibonding σ* orbital and the forming bond.
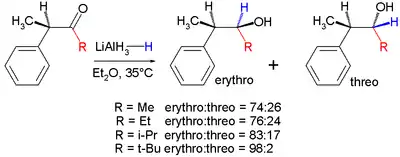
Improving Felkin–Anh selectivity for organometal additions to aldehydes can be achieved by using organo-aluminum nucleophiles instead of the corresponding Grignard or organolithium nucleophiles. Claude Spino and co-workers[26] have demonstrated significant stereoselectivity improvements upon switching from vinylgrignard to vinylalane reagents with a number of chiral aldehydes.
Cram’s rule
Addition of achiral allylmetals to aldehydes forms a chiral alcohol, the stereochemical outcome of this reaction is determined by the chirality of the α-carbon on the aldehyde substrate (Figure "Substrate control: addition of achiral allylmetals to α-chiral aldehydes"). The allylmetal reagents used include boron, tin and titanium.
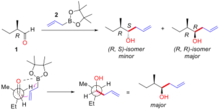
Cram’s rule explains the stereoselectivity by considering the transition state depicted in figure 3. In the transition state the oxygen lone pair is able to interact with the boron centre whilst the allyl group is able to add to the carbon end of the carbonyl group. The steric demand of this transition state is minimized by the α-carbon configuration holding the largest group away from (trans to) the congested carbonyl group and the allylmetal group approaching past the smallest group on the α-carbon centre. In the example below (Figure "An example of substrate controlled addition of achiral allyl-boron to α-chiral aldehyde"), (R)-2-methylbutanal (1) reacts with the allylboron reagent (2) with two possible diastereomers of which the (R, R)-isomer is the major product. The Cram model of this reaction is shown with the carbonyl group placed trans to the ethyl group (the large group) and the allyl boron approaching past the hydrogen (the small group). The structure is shown in Newman projection. In this case the nucleophilic addition reaction happens at the face where the hydrogen (the small group) is, producing the (R, R)-isomer as the major product.
Chiral auxiliaries
Asymmetric stereoinduction can be achieved with the use of chiral auxiliaries. Chiral auxiliaries may be reversibly attached to the substrate, inducing a diastereoselective reaction prior to cleavage, overall producing an enantioselective process. Examples of chiral auxiliaries include, Evans’ chiral oxazolidinone auxiliaries (for asymmetric aldol reactions)[27] pseudoephedrine amides and tert-butanesulfinamide imines.
Substrate control: asymmetric induction by molecular framework in cyclic systems
Cyclic molecules often exist in much more rigid conformations than their linear counterparts. Even very large macrocycles like erythromycin exist in defined geometries despite having many degrees of freedom. Because of these properties, it is often easier to achieve asymmetric induction with macrocyclic substrates rather than linear ones. Early experiments performed by W. Clark Still[28] and colleagues showed that medium- and large-ring organic molecules can provide striking levels of stereo induction as substrates in reactions such as kinetic enolate alkylation, dimethylcuprate addition, and catalytic hydrogenation. Even a single methyl group is often sufficient to bias the diastereomeric outcome of the reaction. These studies, among others, helped challenge the widely-held scientific belief that large rings are too floppy to provide any kind of stereochemical control.
A number of total syntheses have made use of macrocyclic stereocontrol to achieve desired reaction products. In the synthesis of (−)-cladiella-6,11-dien-3-ol,[29] a strained trisubstituted olefin was dihydroxylated diasetereoselectively with N-methylmorpholine N-oxide (NMO) and osmium tetroxide, in the presence of an unstrained olefin. En route to (±)-periplanone B,[30] chemists achieved a facial selective epoxidation of an enone intermediate using tert-butyl hydroperoxide in the presence of two other alkenes. Sodium borohydride reduction of a 10-membered ring enone intermediate en route to the sesquiterpene eucannabinolide[31] proceeded as predicted by molecular modelling calculations that accounted for the lowest energy macrocycle conformation. Substrate-controlled synthetic schemes have many advantages, since they do not require the use of complex asymmetric reagents to achieve selective transformations.
Reagent control: addition of chiral allylmetals to achiral aldehydes
In organic synthesis, reagent control is an approach to selectively forming one stereoisomer out of many, the stereoselectivity is determined by the structure and chirality of the reagent used. When chiral allylmetals are used for nucleophilic addition reaction to achiral aldehydes, the chirality of the newly generated alcohol carbon is determined by the chirality of the allymetal reagents (Figure 1). The chirality of the allymetals usually comes from the asymmetric ligands used. The metals in the allylmetal reagents include boron, tin, titanium, silicon, etc.
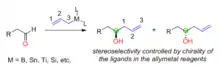
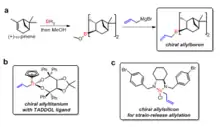
Various chiral ligands have been developed to prepare chiral allylmetals for the reaction with aldehydes. H. C. Brown was the first to report the chiral allylboron reagents for asymmetric allylation reactions with aldehydes.[32] The chiral allylboron reagents were synthesized from the natural product (+)-a-pinene in two steps. The TADDOL ligands developed by Dieter Seebach has been used to prepare chiral allyltitanium compounds for asymmetric allylation with aldehydes.[33] Jim Leighton has developed chiral allysilicon compounds in which the release of ring strain facilitated the stereoselective allylation reaction, 95% to 98% enantiomeric excess could be achieved for a range of achiral aldehydes.[34]
See also
References
- IUPAC Gold Book definition Link
- Asymmetric Synthesis of Natural Products, Ari Koskinen ISBN 0-471-93848-3
- Studies in Stereochemistry. X. The Rule of "Steric Control of Asymmetric Induction" in the Syntheses of Acyclic Systems Donald J. Cram, Fathy Ahmed Abd Elhafez J. Am. Chem. Soc.; 1952; 74(23); 5828–5835. Abstract
- Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones Marc Chérest, Hugh Felkin and Nicole Prudent Tetrahedron Letters Volume 9, Issue 18, 1968, Pages 2199-2204 doi:10.1016/S0040-4039(00)89719-1
- It bears mentioning that in Vietnamese, the surname is given first, and so this would be better called the Felkin–Nguyen Model.
- Anh, N. T.; Eisenstein, O. Nouv. J. Chim. 1977, 1, 61.
- Anh, N. T.; Eisenstein, O.; Lefour, J-M.; Dau, M-E. J. Am. Chem. Soc. 1973, 95, 6146.
- Bürgi, H. B.; Dunitz, J. D.; Shefter, E. J. Am. Chem. Soc. 1973, 95, 5065.
- Bürgi, H. B.; Dunitz, J. D.; Lehn, J. M.; Wipff, G. Tetrahedron 1974, 30, 1563.
- Anh, N. T.; Eisenstein, O. Tetrahedron Lett. 1976, 155.
- Anh, N. T. Top. Curr. Chem. 1980, 88, 146.
- Mengel A., Reiser O.Chem. Rev., 1999, 99 (5), 1191–1224.
- Cram DJ, Elhafez FA. J. Am. Chem. Soc.; 1952; 74(23); 5828–5835.
- Reetz MT, Hullmann M, Seitz T. Angew. Chem. Int. Ed. Engl. 1987. 26, 477–480.
- Cornforth JW, Cornforth MRH, Mathew KK. J. Chem.Soc. 1959, 112–127.
- Cherest M, Felkin H, Prudent N. Tetrahedron Lett. 1968, 18, 2199–2204.
- Reetz, M.T.; Jung, A. J. Am. Chem. Soc., 1983, 105, 4833.
- Leitereg, T.J.; Cram, D.J. J. Am. Chem. Soc. 1968, 90, 4011.
- Reetz. M.T.; Kesseler, K.; Jung, A. Tetrahedron Lett. 1984, 25, 729.
- Evans, D.A.; Duffy, J.L.; Dart, M.J. Tetrahedron Lett. 1994, 35, 8537.
- Evans, D.A.; Dart, M.J.; Duffy, J.L.; Yang, M.G. J .Am. Chem. Soc. 1996, 118, 4322.
- Clayden; Greeves; Warren; Wothers (2001). Organic Chemistry. Oxford University Press. p. 895. ISBN 978-0-19-850346-0.
- Houk, K. N. et al., Science, 1986, 231, 1108-1117.
- a) Anh, N. T. Top. Curr. Chem. 1980, 88, 145–162; (b) Anh, N. T.; Eisenstein, O. Nouv. J. Chim. 1977, 1, 61–70; (c) Anh, N. T.; Eisenstein, O. Tetrahedron Lett. 1976, 26, 155–158.
- Burgi, H. B.; Dunitz, J. D.; Lehn, J. M.; Wipff, G. Tetrahedron. 1974. 12, 1563–1572.
- Spino, C.; Granger, M. C.; Boisvert, L.; Beaulieu, C. Tetrahedron Lett. 2002, 43, 4183–4185.
- Evans, D. A.; Bartroli, J.; Shih, T. L., Am. Chem. Soc., 1981, 103, 2127-2129.
- Still, W. C.; Galynker, I. Tetrahedron 1981, 37, 3981-3996.
- Kim, Hyoungsu; Lee, Hyunjoo; Kim, Jayoung; Kim, Sanghee; Kim, Deukjoon (2006-12-01). "A General Strategy for Synthesis of Both (6Z)- and (6E)-Cladiellin Diterpenes: Total Syntheses of (−)-Cladiella-6,11-dien-3-ol, (+)-Polyanthellin A, (−)-Cladiell-11-ene-3,6,7-triol, and (−)-Deacetoxyalcyonin Acetate". Journal of the American Chemical Society. 128 (49): 15851–15855. doi:10.1021/ja065782w. ISSN 0002-7863. PMID 17147397.
- Still, W. Clark (1979-04-01). "(.+-.)-Periplanone-B. Total synthesis and structure of the sex excitant pheromone of the American cockroach". Journal of the American Chemical Society. 101 (9): 2493–2495. doi:10.1021/ja00503a048. ISSN 0002-7863.
- Still, W. Clark; Murata, Shizuaki; Revial, Gilbert; Yoshihara, Kazuo (1983-02-01). "Synthesis of the cytotoxic germacranolide eucannabinolide". Journal of the American Chemical Society. 105 (3): 625–627. doi:10.1021/ja00341a055. ISSN 0002-7863.
- Brown, H. C.; Jadhav, P. K. J. Am. Chem. Soc. 1983, 105, 2092.
- Duthaler, R. O.; Hafner, A. Chem. Rev. 1992, 92, 807.
- Kinnaird, J. W. A.; Ng, P. Y.; Kubota, K.; Wang, X.; Leighton, J. L. J. Am. Chem. Soc. 2002, 124, 7920.
External links
- The Evolution of Models for Carbonyl Addition Evans Group Afternoon Seminar Sarah Siska February 9, 2001