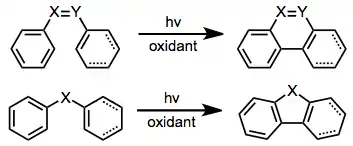
Mallory reaction
In organic chemistry, the Mallory reaction is a photochemical-cyclization–elimination reaction of diaryl-ethylene structures to form phenanthrenes and other polycyclic form polycyclic aromatic hydrocarbons and heteroaromatics.[1][2] This name reaction is named for Frank Mallory, who discovered it while a graduate student.[3]
Under UV irradiation, stilbene and its derivatives undergo intramolecular cyclization to form dihydrophenanthrenes. In the presence of an oxidant, the dihydrophenanthrenes aromatize to give polycyclic aromatics. Typically, the dihydrophenanthrenes themselves are relatively unstable, and revert to cis-stilbenes in the absence of a hydrogen-trapping agent. Suitably substituted stilbenes may undergo irreversible, rearomatizing elimination or [1,n]-shift processes in the absence of an oxidant. Aryl enynes,[4] heteroatomic stilbene derivatives (e.g. amides[5]), and substrates containing a single heteroatom in place of the stilbene double bond[6] also undergo the reaction.
Mechanism and stereochemistry
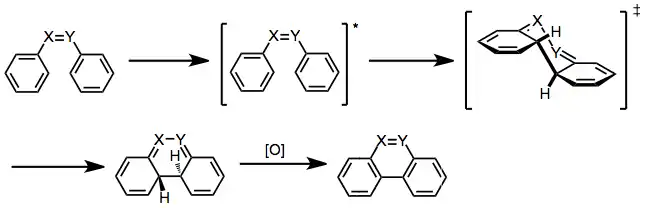
Regardless of the presence or absence of an oxidant, the first step of the reaction is photochemical excitation of a stilbene or similar structure, leading to formation of a dihydrophenanthrene or similar intermediate. For stilbene and other chemicals containing a double-bond linker between the two aromatic rings, the excited structure can undergo reversible cis-trans isomerization. Although only cis structures can undergo the cyclization step themselves, trans structures can isomerize in situ and then cyclize.[2] In keeping with the Woodward–Hoffmann rules, molecular orbital symmetry analysis of the photochemical reaction of the six-electron system explains the trans relative configuration at the newly bound centers by a conrotatory process.[7]
This cyclization is reversible, but several other subsequent reactions can occur instead, depending on structural details and whether certain other reagents are present.
Oxidative conditions
In the presence of an oxidizing agent, the cyclized intermediate can be oxidized to aromatize the rings. For example, dihydrophenanthrene becomes phenanthrene. Oxygen and iodine are the most commonly employed oxidants.
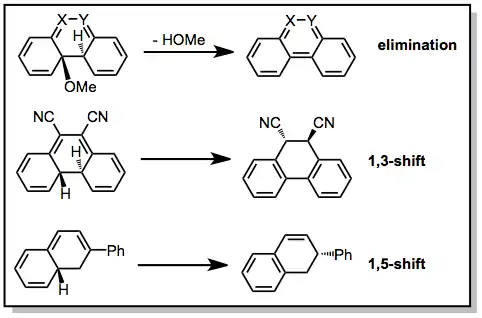
Non-oxidative conditions
For most substrates, in the absence of an oxidant, the dihydrophenanthrene intermediate may reversibly open to the corresponding cis-stilbene. However, suitably substituted stilbenes cyclize irreversibly if an aromatizing elimination or hydrogen shift process can take place. Examples of these transformations are provided below.[8][9][10]
Scope and limitations
Photocyclization can be carried out with ortho-, meta-, and para-substituted stilbene substrates. ortho-Substituted substrates generally give 1-substituted phenanthrenes, unless the substituent is a good leaving group, in which case elimination to form unsubstituted phenanthrene occurs.[11] meta- Substituted substrates give mixtures of 2- and 4-substituted products.
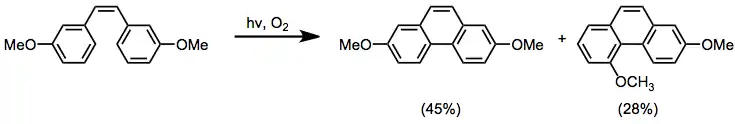
Substitution of the exocyclic double bond is well tolerated. Polycyclic aromatic compounds can be synthesized using substrates containing multiple aromatic rings.[9]
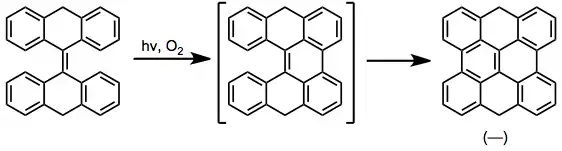
Stilbene derivatives containing fused aromatic systems may cyclize using either of two nonequivalent ortho carbons. Which carbon reacts depends on both steric and electronic factors. Electronically, the dihydrophenanthrene intermediate exhibiting greater aromatic stabilization is preferred. For instance, in 1-naphthyl-2-phenylethylene, electronic factors favor the formation of 1 over 2 in a ratio of 98.5:1.5.[12]
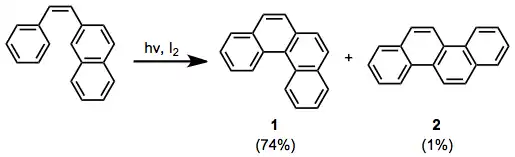
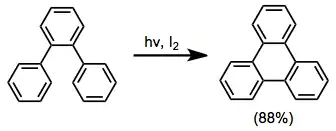
ortho-Terphenyl substrates cyclize to the corresponding triphenylenes in the presence of an oxidant, such as iodine. Oxygen is unsatisfactory because ring-opening to highly stabilized terphenyl is faster than oxidation when oxygen is used.[13]
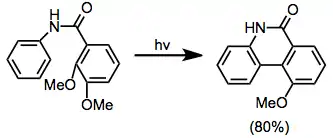
Amides may cyclize to form lactams. Esters, which exist primarily in the trans conformation about the C-O single bond, do not undergo this process efficiently.[14]
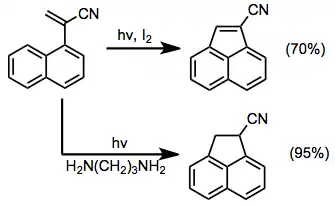
Photocyclization can also form five-membered rings. In the vinyl naphthalene series, both oxidative[10] and non-oxidative processes are possible; although the latter requires a proton-transfer catalyst.[15]
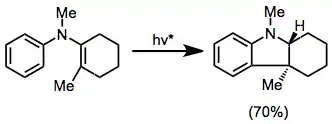
Cyclization of arylvinyl- or diarylamines provides indolines and carbazoles, respectively. In one interesting example, the use of circularly polarized light provided 3 in slight enantiomeric excess.[16]
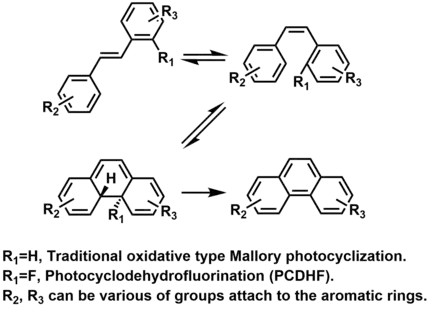
In 2015, Li and Twieg reported a novel derivative of Mallory type photocyclizations and named it as photocyclodehydrofluorination (PCDHF). In the cyclization a stilbene (or ortho-terphenyl) with a pentafluorophenyl group, the fluorine atom can be used as a facile leaving group.[17]
Synthetic applications
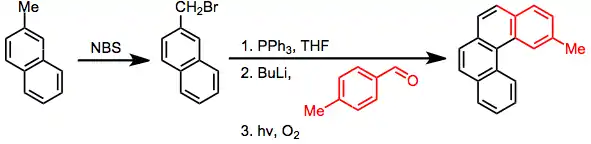
Photocyclization can be used as the final step of a sequence to generate a fused aromatic ring at a benzylic position. After benzylic bromization with N-bromosuccinimide, transformation to the phosphonium salt, and a Wittig reaction with anaromatic aldehyde, photocyclization fuses the aromatic rings. Iteration of this sequence results in helicenes.[18]
See also
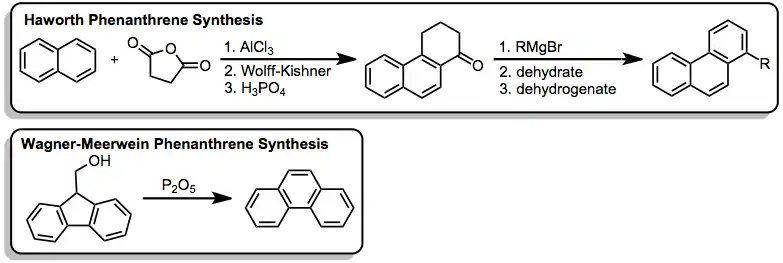
Several other methods are available to synthesize the phenanthrene ring system; however, most of these are longer or less functional group tolerant than photocyclization. The Haworth reaction and the Wagner-Meerwein-type ring-expansion are two such alternatives.[19]
References
- Lvov, A. G. J. Org. Chem. 2020, 85, 8749–8759. doi:10.1021/acs.joc.0c00924
- Mallory, F. B.; Mallory, C. W. Org. React. 1984, 30, 1. doi:10.1002/0471264180.or030.01
- Cassidy, Kim. (November 16, 2017) "Remembering the Distinguished Career of Long-Time Professor of Chemistry Frank Mallory"
- Tinnemans, A. H. A.; Laarhoven, W. H. J. Chem. Soc., Perkin Trans. 1, 1976, 1115.
- Thyagarajan, B. S.; Kharasch, N.; Lewis, H. B.; Wolf, W. Chem. Commun., 1967, 614.
- Zeller, K.-P.; Petersen, H. Synthesis 1975, 532.
- Cuppen, J. H. M.; Laarhoven, W. H. J. Am. Chem. Soc. 1972, 94, 5914.
- Giles, R. G. F.; Sargent, M. V. J. Chem. Soc., Perkin Trans. 1, 1974, 2447.
- Sargent, M. V.; Timmons, C. J. J. Chem. Soc. Suppl. 1, 1964, 5544.
- Lapouyade, R.; Koussini, R.; Rayez, J.-C. J. Chem. Soc., Chem. Commun., 1975, 676.
- Cava, M. P.; Stern, P.; Wakisaka, K. Tetrahedron, 29, 2245 (1973).
- Mallory, F. B.; Mallory, C. W.; Halpern, E. J. First Middle Atlantic Regional Meeting of the American Chemical Society, February 3, 1966, Philadelphia, Pa., Abstracts, p. 134.
- Sato, T.; Shimada, S.; Hata, K. Bull. Chem. Soc. Jpn. 1971, 44, 2484.
- Ninomiya, I.; Naito, T.; Kiguchi, T. J. Chem. Soc., Perkin Trans. 1, 1973, 2257.
- Lapouyade, R.; Koussini, R.; Bouas-Laurent, H. J. Am. Chem. Soc. 1977, 99, 7374.
- Nicoud, J. F.; Kagan, H. B. Isr. J. Chem. 1977, 15, 78.
- Li, Zhe; Twieg, Robert J. (2015-09-11). "Photocyclodehydrofluorination". Chemistry - A European Journal. 21 (44): 15534–15539. doi:10.1002/chem.201502473. ISSN 0947-6539. PMID 26360126.
- Laarhoven, W. H.; Cuppen, Th. J. H. M.; Nivard, R. J. F. Tetrahedron 1974, 30, 3343.
- Floyd, A. J.; Dyke, S. F.; Ward, S. E. Chem. Rev. 1976, 76, 509. doi:10.1021/cr60303a001