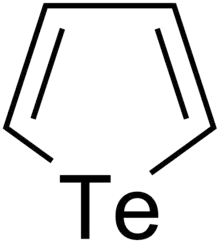
Tellurophenes
Tellurophenes are the tellurium analogue of thiophenes and selenophenes.
 | |||
| |||
| Names | |||
|---|---|---|---|
| Preferred IUPAC name
Tellurophene[1] | |||
| Identifiers | |||
3D model (JSmol) |
|||
| 103225 | |||
| ChEBI | |||
| ChemSpider | |||
| 647889 | |||
PubChem CID |
|||
| |||
| |||
| Properties | |||
| C4H4Te | |||
| Molar mass | 179.68 g·mol−1 | ||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |||
Synthesis
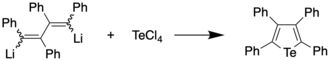
The first preparation of a tellurophene, tetraphenyltellurophene, was reported in 1961 by Braye et al.[2][3] by reacting 1,4-dilithiotetraphenylbutadiene with tellurium tetrachloride, with the former synthesized by reaction of diphenylacetylene and lithium metal. The tellurophene, upon recrystallization from a dichloromethane/ethanol mixture, was obtained in 56% yield, and found to appear as yellow-orange crystals with a melting point of 239-239.5 °C. The same compound was obtained from 1,4-diiodotetraphenylbutadiene and lithium telluride in 82% yield.[2][4]
In 1966, Mack report a synthesis of an unsubstituted tellurophene through the reaction of sodium telluride with diacetylene in methanol at 20 °C. This method could be generalised to prepare 2,5-derivatives of tellurophene by selecting a suitably-substituted diacetylene precursor.[5] The product was obtained as a pale yellow liquid with a melting and boiling point of −36 °C and 148 °C, respectively. Taticchi et al. improved upon this synthesis by using a Schlenk line to exclude oxygen and moisture from the reaction vessel, using pure butadiyne (to decrease unwanted oxidation and polymerization side reactions), and by not using a vacuum to remove the methanol as it leads to loss of the product. This improved procedure allowed the tellurophene to be isolated in 47% yield.[4][6]
The geometry of tellurophene was first determined in 1973 through microwave spectroscopy, and has been further refined through X-ray diffraction studies.[7] It has been found that the Te–C bond has a length of 2.046 Å, which is longer than that of selenophene. Further, the C–Te–C angle has been determined to be 82°, smaller than that found in selenophene, an observation attributed to the larger size of the tellurium atom. These findings are also consistent with the aromaticity of selenophene being greater than that of tellurophene; amongst its congeners, the order of decreasing aromaticity has been demonstrated to be: benzene > thiophene > selenophene > tellurophene > furan.[4][8]
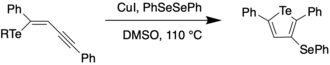
A variety of protocols for the synthesis of tellurophenes have been developed, such as metal-catalyzed cross coupling reactions and cyclization of enynes.[9][10] Some examples are shown below. In 2008, Zeni et al. reported on the copper-catalyzed cyclizations of chalcogenoenynes to obtain 3-substituted chalcogenophenes which could be further functionalized using boronic acids via palladium-catalyzed Suzuki coupling.[9]
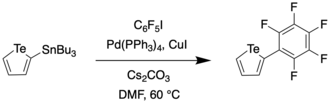
In 2016, Taylor et al. reported a synthetic route to a perfluoroaryl substituted tellurophene through Stille coupling.[10] This compound was then subjected to further iodination and sequential Sonogashira couplings to generate a receptor for anions such as Cl− and Br−.
However, metal-catalyzed cross-coupling reactions to synthesize 3-functionalized tellurophenes were deemed to be cumbersome as they required 3-bromo- or 3-iodo-tellurophenes, the syntheses of which could be quite complicated.[11] An alternative method was reported by Seferos et al. in 2013,[12] but this method was hindered by low yields and the use of expensive starting materials such as the Weinreb amide.
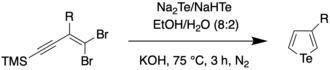
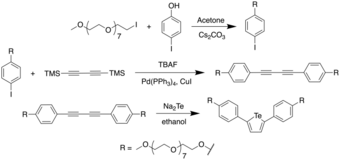
In 2018, Han et al. reported on a one-pot procedure for the synthesis of a variety of functionalized tellurophenes without the use of transition metals.[11] This was done by reacting substituted 1,1-dibromo-1-en-3-ynes with telluride salts (Na2Te/Na2Se) under mild conditions. The telluride salts were synthesized through an earlier protocol, wherein Te/Se was reduced with sodium borohydride in ethanol.[12] The synthesis of the 3-functionalized tellurophenes is as follows:
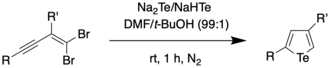
It was found through mechanistic studies that the reaction was highly influenced by the polarity of the solvent. Polar solvents such as water were thought to polarize the Te–H bond, thus increasing the negative charge on Te and making it more nucleophilic. To obtain a wider scope of the reaction, the authors used dimethylformamide (DMF) as the solvent since DMF not only has a higher dielectric constant (and therefore, higher polarity) than water, but also was found to be able to dissolve enynes better compared to water. Using a solvent combination of DMF and t-BuOH, the authors were able to synthesize 2,4-disubstituted tellurophenes at room temperature.
Reactivity
Anion receptor
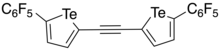
In 2016, Taylor et al. developed a bidentate and electron-deficient bistellurophene receptor in which the tellurophenes were linked through an ethynylene bridge.[10] As the tellurophene was thought to function as a Lewis acid in its interaction with the anion (Lewis base), 2,5-diaryltellurophenes with electron-deficient arene substituents were synthesized. Through monitoring the change in the optical absorption spectrum upon addition of Bu4N+Cl− in tetrahydrofuran (THF), it was found that 2,5-bis[(perfluoro)aryl]tellurophene was able to bind Cl− with an association constant (Ka) of 310 ± 20 L mol−1, and would also bind Br− and BzO−. Using computational studies, it was found that an ethynylene linkage between two tellurophenes would place the chalcogen bond donors at an appropriate distance such that the receptor could form two chalcogen bonds with the chloride. Through sequential Sonogashira coupling reactions, an ethynylene-linked bistellurophene was synthesized from 2-iodo-5-(perfluorophenyl)tellurophene. Upon addition of [Bu4N]Cl to a solution of the receptor in THF, changes to the absorption spectrum were found showing a Ka = 2290 L mol−1. The significantly higher Ka was found to be in agreement with density functional theory calculations which showed that the minimum-energy geometry was one where the chloride anion was in between the tellurium atoms, with Te–Cl bond distances of 3.23 Å and Cl–Te–C angles of 170°. One significant difference of the bidentate receptor was that there was no anion-arene stabilizing influence, and operated through purely chalcogen bonding, unlike the monodentate receptor.
Halogen photoelimination
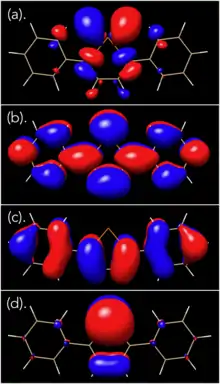
In 2013, Seferos et al. reported the first example of photoreductive elimination (PE) of Cl2 and Br2 from an isoindigo-substituted tellurophene, 2,5-bis[5-(N,N′-dihexylisoindigo)]tellurophene.[18] Due to the extensive π-conjugation which resulted in low-energy absorption, relatively low-energy light (505 nm) was used to photoexcite the halogenated species to drive the photoreductive elimination (PE). However, the quantum yields for PE of Cl2 and Br2 were found to be 0.19% and 0.18%, respectively. Through DFT calculations, it was found that the main transition upon photoexcitation was a HOMO to LUMO+2 transition at 535 nm, with the LUMO+2 state possessing Te-X antibonding character. It was postulated that the low quantum yields were due to the fact that there were no lower energy excited states with Te-X antibonding character, and that this would limit the efficiency of the reaction. Therefore, it was thought that by changing the substituents on tellurophene such that the main transition upon photoexcitation would be HOMO to LUMO, this would significantly improve the reaction by removing efficiency losses through relaxations from states that did not possess Te-X antibonding character and did not promote Te-X bond dissociation.
In 2015, Seferos et al. demonstrated that 2,5-diphenyltellurophene (PT) could participate in photoreductive elimination of fluorine, chlorine, and bromine via the two-electron Te(IV)/Te(II) photocycle, with quantum yields of up to 16.9%.[19] This was the first report of an organotellurium compound that could perform photoreductive defluorination. The HOMO and LUMO orbitals of 2,5-diphenyltellurophene were calculated through DFT using the computational program GAMESS, and it shows that the LUMO is delocalized over the entire molecule, in agreement with the orbital pictures reported by Seferos et al.[19] Using DFT calculations using the B3LYP functional, the authors was found that the two strongest optical transitions for PT was the HOMO to LUMO and HOMO to LUMO+1 transitions. Upon addition of halogen, however, it was found that the HOMO-LUMO energy gap decreased, with the LUMO possessing significant Te-X antibonding character. Because of this, it was postulated that by filling the π* orbital with electrons, this would facilitate breaking of the Te-X bond, and hence, halogen dissociation. And indeed, upon addition of excess halogen, the peak at 342 nm corresponding to the tellurophene decreased, while a red-shifted absorption peak appeared, with the peak being more red-shifted as one moved towards the heavier halogens (PT-F2: λmax = 395 nm, PT-Cl2: λmax = 416 nm, PT-Br2: λmax = 433 nm). Upon irradiation of the PT-Br2 sample with a 447.5 nm lamp, it was found that the absorption spectrum of the sample rapidly changed back to that of PT in 12 seconds. This was also observed using 1H NMR spectroscopy. With F2, however, it was found that there were significant decomposition products owing to the halogen's high reactivity towards PT. This was circumvented by using water as a halogen trap instead of DMBD (2,3-dimethyl-1,3-butadiene), since fluorine exhibits a high reactivity in water to form hydrofluoric acid.
Photooxidation
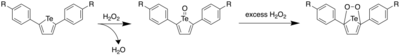
In 2013, Seferos et al. reported the first example of a water-soluble tellurophene by attaching octaethylene glycol monomethyl ether (OEG) substituents on the para-position of the phenyl groups on 2,5-diphenyltellurophene.[20] This was done by first synthesizing iodo-OEG, which was then added to 4-iodophenol to form iodo-4-OEG-benzene. This was then subjected to a Sonogashira coupling, and the resulting butadiyne was treated with sodium telluride, producing the desired product. Treating the tellurophene with hydrogen peroxide (H2O2) resulted in a red-shifted absorption peak at 435 nm in the UV-vis spectrum, with a peak appearing at 280 nm with a concomitant decrease of the peak at 435 nm upon treatment with excess peroxide. Through studying the reaction rate, it was found that the reaction was first order in both H2O2 and tellurophene. It was also found that the product with absorption peak at 435 nm was a dihydroxytellurophene, and the product at 280 nm was a telluroketone. The telluroketone was also found to be generated upon irradiation of a solution of the tellurophene in water with blue LED light, showing that it could be oxidized by singlet oxygen. Further, by treating a solution of the telluroxide to a -0.5 V potential (vs. Ag/AgCl), it was found that its absorption peak decreased with the concurrent increase of the peak at 354 nm corresponding to the diaryltellurophene. This process could be reversed upon applying a potential of 0.8 V, thus indicating reversible oxidation.
In 2017, Seferos et al. reported the oxidative ring opening of 2,5-diphenyltellurophene (PT) under aerobic conditions, and with meta-chloroperoxybenzoic acid (mCPBA).[21] Through DFT calculations, it was found that upon oxidation of PT, the resulting Te(IV) oxide PT-O had a lower HOMO level, with the LUMO being significantly stabilized. This led to a large decrease in the HOMO-LUMO energy gap, which predicted a red shift in the maxima of the absorption spectrum. Furthermore, it was found that the electron density of the LUMO included a Te-O σ* orbital. This orbital picture has been further reproduced using Avogadro and GAMESS, as shown on the orbital diagram below.[16][17]
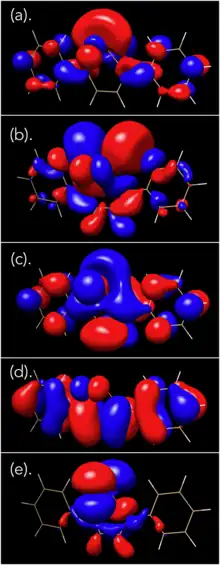
Addition of 1 equiv of mCPBA to a solution of PT led to an immediate colour change from colourless to yellow. However, upon adding more mCPBA (4 equiv), there was a gradual decrease in the absorbance at 388 nm and a resulting absorption increase below 300 nm. As one would expect a red shift based on computational calculations, the observed blue shift suggested that upon formation of telluroxide, a different reaction pathway prevented the formation of the tellurone (PT-O2). By analyzing the reaction by 1H NMR spectroscopy, it was found that the yellow solid that had formed as the product had a downfield shift at 1123.3 ppm upon addition of mCPBA. NMR spectra and the absorption spectrum of the product in solution led to the authors attributing this product as the telluroxide. Upon addition of a large excess of mCPBA (8 equiv), the solution became bright yellow, which slowly diminished upon stirring overnight. The final product was an insoluble white solid (TeO2) and a colorless solid which was found to be (Z)-1,4-diphenylbut-2-ene1,4-dione, (Z)-ED, through a combination of mass spectrometry and 1H NMR data. This result confirmed that the tellurone is not formed even after addition of excess mCPBA.
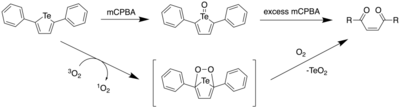
By using a singlet oxygen trap, 9,10-diphenylanthracene, the authors investigated the formation of singlet oxygen upon irradiation of PT. Upon irradiation of a solution containing both PT and 9,10-DPA with white light, a decrease in the absorbance at 355 nm was observed, which was indicative of 1O2 formation since 9,10-diphenylanthracene undergoes 1,4-addition with 1O2 to form the endoperoxide. A solution of PT in CDCl3 was then irradiated with 365 nm light, and it was observed that after 1 hour, complete conversion from PT to (Z)-ED had occurred with the concomitant formation of TeO2.
It had been reported by Nakayama et al. that addition of 4 equiv of mCPBA to a tetraphenylselenophene solution also resulted in the formation of ene-dione compounds and SeO2.[22] The proposed mechanism was where the first equivalent of mCPBA forms the selenoxide, with the extra three equivalents reacting with selenophene to produce a selenone diepoxide intermediate. This mechanism was consistent with the formation of telluroxide upon addition of mCPBA, and the formation of an ene-dione product upon addition of 4 equivalents of mCPBA.
Optoelectronic properties
Compared to thiophenes, tellurophenes have been found to have lower optical band gaps, significantly lower LUMO levels, and higher charge carrier mobilities.[23] In 2014, Rivard et al. reported the phosphorescence of pinacolboronate-substituted tellurophenes at room temperature,[24] in contrast to previously reported phosphorescent materials made of expensive rare metals such as iridium and platinum. The phosphorescence was found to be aggregation-induced, as the tellurophene was non-emissive when dissolved in THF, but glowed bright green in the solid state and upon aggregation in THF/water solutions. A dibrominated Te(IV) tellurophene, B-TeBr2-B, was found to be non-emissive, indicating that the Te(II) center in B-Te-B plays an important role in phosphorescence. By replacing the pinacolboronate esters with thiophenes, there was no luminescence, indicating that both Te(II) and BPin played a cooperative role leading to emission. DFT calculations on B-Te-B revealed that the HOMO has significant contribution from the lone pair on the Te p-orbital, with the LUMO being significantly delocalized over the B-C bond. Furthermore, the energy of the triplet state (T3) was found to be degenerate with the singlet excited state (S1), which was proposed to lead to efficient singlet-triplet crossing to occur, leading to emission.[25] This was in contrast to the sulfur and selenium analogues, where the triplet state was found to be ~1 eV higher in energy.
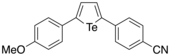
In 2018, Okuma et al. reported the synthesis of various 2,5-diaryltellurophenes substituted with electron-donating and electron-withdrawing groups through sequential ditelluride exchange and intramolecular cyclization reactions.[26] By having both electron-donating (e.g. OMe) and electron-withdrawing (e.g. CN) groups on the tellurophene simultaneously, this resulted in a sharp reduction of the HOMO-LUMO gap. Furthermore, the authors observed significant solvatochromism, since the emission maxima shifted to longer wavelengths with increasing solvent polarity. Through DFT calculations, the authors was found that the HOMO was localized on the π-orbitals of the tellurophene and the electron-donating substituent, with the LUMO localized over the π*-orbitals of the tellurophene and the electron-withdrawing substituent. It was concluded that having both electron-donating and electron-withdrawing substituents stabilizes the LUMO, with the HOMO-LUMO transitions having significant charge-transfer character, which in turn explained the solvatochromic effect. This work therefore showed how one can tune the optoelectronic properties of π-conjugated tellurophenes.
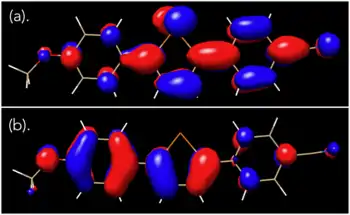
The same molecule was subjected to DFT calculations using the computational program GAMESS, where it was found that the HOMO and LUMO orbitals were in qualitative agreement with the orbital pictures reported by Okuma, showing that the HOMO and LUMO show extensive orbital delocalization on the p-anisyl and p-cyanophenyl substituents, respectively.
Polymers
In 2016, Seferos et al. reported the synthesis of well-defined, high-molecular-weight poly-3-alkyltellurophenes (P3ATe) through catalyst transfer polymerization (CTP).[27][28] CTP is an important route to synthesize polymers with a narrow molecular weight distribution and a well defined end-group,[29] but it was found in 2013 that applying CTP-conditions for the synthesis of P3ATe led to polymers with low molecular weights, and broad polydispersities. To obtain P3ATe with a narrow polydispersity, the authors investigated the optimal conditions using kinetic studies and DFT calculations. It was found experimentally that the branched side chain played an important role on the polymerization rate and polymer quality. To mitigate this effect, monomers with various other side chains were synthesized. From this, it was found that moving the ethyl branches away from the heterocycle to the more remote 3- and 4- positions led to an improved polymerization rate and control, such that P3ATe with narrow polydispersities and high molecular weights were obtained. This improvement was attributed to the lack of steric hindrance. Furthermore, it was found that upon moving the branching point away from the heterocycle led to a red-shift in the optical absorption, which was attributed to a decrease in the degree of twisting, resulting in an increase in the conjugation between the tellurophene backbone.
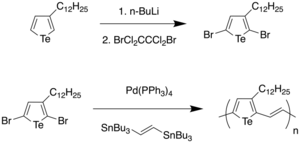
Heeney et al. reported the synthesis of the first tellurophene-vinylene copolymer through Stille coupling of 2,5-dibromo-3-dodecyltellurophene and (E)-1,2-bis(tributylstannyl)ethylene, resulting in P3TeV in 57% yield with an approximate Mn of 10 kDa and a polydispersity of 2.4.[30] By synthesizing thiophene and selenophene analogues, it was found that there was a reduction in the optical band gap as a result of the stabilization of the LUMO, resulting in a small band gap of 1.4 eV for P3TeV. By constructing organic field effect transistors (OFETs), it was found that the selenophene polymer had the highest charge mobility, and that the tellurium analogue did not lead to an increase in mobility despite the larger size of tellurium, and possibility of closer interchain Te-Te interactions, which was attributed to the low solubility of P3TeV which resulted in poor film formation. Therefore, the authors remarked that future work entailed modifying the side-chains to increase solubility.
Frustrated Lewis Pair chemistry
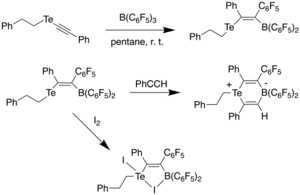
In 2015, Stephan et al. reported a vinyl telluroether with a pendant borane which acted as an intramolecular frustrated Lewis pair (FLP).[31] This was achieved by reacting tellurium acetylide with B(C6F5)3 in pentane at room temperature, affording bright orange crystals in 94% yield. It was found through 11B NMR that the product had a four-coordinate boron, which indicated a weak Te-B interaction due to the broad signal. Reacting this compound with phenylacetylene at room temperature resulted in a cis-1,2-addition across the alkyne bond, generating a zwitterionic, six-membered Te-B heterocycle, as observed using X-ray diffraction spectroscopy. It was found that the coordination geometries around tellurium and boron were pseudo-trigonal pyramidal, and pseudo-tetrahedral, respectively. The C-C bond length was observed to be 1.326(4) Å, much longer than a C-C triple bond and closer to a C-C double bond, indicating that the compound had activated phenylacetylene through FLP chemistry. However, unlike other reported FLP compounds,[32] it was unable to activate H2 or bind CO2, which was attributed to the fact that telluroethers are poor nucleophiles. Although the telluroether did not undergo oxidation by halogens to produce the corresponding Te(IV) dihalide compounds, it was found to react with iodine to afford a five-membered Te-B-I heterocycle.
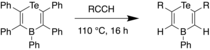
Later, Stephan et al. reported the synthesis of various Te-B heterocycles through reaction of 1-bora-4-tellurocyclohexa-2,5-diene and two equivalents of a terminal alkyne upon heating, with loss of a diarylalkyne.[33] X-ray crystal studies revealed that the C-C bond distances in the heterocycle were close to that of C-C double bonds, indicating delocalization within the molecule. The reaction was proceeded with high regioselectivity, with the terminal CH carbon of the alkyne attaching to the boron. Consistent with this observation, it was proposed that the Te and B act as a FLP, undergoing a [4+2] cyclcoaddition to the alkyne such that the boron adds to the CH carbon, due to the steric bulk at the boron center. In 2018, this FLP chemistry was developed further through the synthesis of 4H-1,4-telluraborine, which was found to be a useful hydroboration reagent for alkenes, ketones, and aldehydes.[34]
Hydrogen-bonded Organic Frameworks (HOFs)
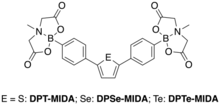
Hydrogen-bonded organic frameworks (HOFs) are porous organic materials that are connected by non-covalent interactions such as hydrogen bonds and π-π interactions. However, due to the relatively weak strength of hydrogen bonds, HOFs rarely exhibit permanent porosity upon removal of solvent molecules.[36] Nonetheless, the weak interactions in HOFs allow the formation of single crystals, which are more amenable to crystallographic studies compared to COFs. Second, HOFs can be easily regenerated through dissolution and recrystallization due to their weak interactions.[36] In 2016, Seferos et al. reported the synthesis of HOFs from chalcogen heterocycles capped with N-methyliminodiacetic acid (MIDA) boronates which contain both hydrogen bond donors and acceptors.[35] MIDA-capped thiophene, selenophene, and tellurophenes were synthesized through Stille coupling. Crystal structures of DPT-MIDA and DPSe-MIDA showed the presence of C-H⋯O hydrogen bonding and C-H⋯π interactions. DPTe-MIDA was not amenable to crystallographic analysis, and it was found through powder X-ray diffraction (PXRD) that DPTe-MIDA had lower crystallinity compared to DPT-MIDA and DPSe-MIDA. However, the main diffraction peaks of DPTe-MIDA were similar to those of DPT-MIDA and DPSe-MIDA, suggesting that all three frameworks self-assembled into similar structures. Thermogravimetric analysis (TGA) revealed that acetonitrile molecules are removed at around 150 °C for DPT-MIDA and DPSe-MIDA, and 70 °C for DPTe-MIDA, with all three HOFs decomposing above 350 °C. DPT-MIDA had the highest surface area, as found by CO2 adsorption at 0 °C. Furthermore, it was found that DPT-MIDA and DPSe-MIDA adsorbed 1 mol of CO2 per mol of building block, whereas DPTe-MIDA adsorbed 0.5 mol of CO2. Furthermore, it was observed that DPTe-MIDA exhibited weak fluorescence compared to DPT-MIDA, which had a quantum yield of 6.6%.
References
- International Union of Pure and Applied Chemistry (2014). Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013. The Royal Society of Chemistry. p. 883. doi:10.1039/9781849733069. ISBN 978-0-85404-182-4.
- Braye, E. H.; Hübel, W.; Caplier, I. (1961). "New Unsaturated Heterocyclic Systems. I". Journal of the American Chemical Society. 83 (21): 4406–4413. doi:10.1021/ja01482a026.
- Rhoden, Cristiano R. B.; Zeni, Gilson (2011). "New development of synthesis and reactivity of seleno- and tellurophenes". Organic & Biomolecular Chemistry. 9 (5): 1301–1313. doi:10.1039/c0ob00557f. ISSN 1477-0520. PMID 21210032.
- Fringuelli, Francesco; Marino, Gianlorenzo; Taticchi, Aldo (1977). Tellurophene and Related Compounds. Advances in Heterocyclic Chemistry. Vol. 21. pp. 119–173. doi:10.1016/S0065-2725(08)60731-X. ISBN 9780120206216.
- Mack, W. (1966). "Synthesis of Tellurophene and its 2,5-Disubstituted Derivatives". Angew. Chem. Int. Ed. 5 (10): 896. doi:10.1002/anie.196608961.
- Fringuelli, Francesco; Taticchi, Aldo (1972). "Tellurophen and some of its derivatives". Journal of the Chemical Society, Perkin Transactions 1: 199–203. doi:10.1039/P19720000199.
- Lukevics, E.; Arsenyan, P.; Belyakov, S.; Pudova, O. (2002). "Molecular Structure of Selenophenes and Tellurophenes". Chemistry of Heterocyclic Compounds. 38 (7): 763–777. doi:10.1023/a:1020607300418. ISSN 0009-3122. S2CID 92305752.
- Fringuelli, Francesco; Marino, Gianlorenzo; Taticchi, Aldo; Grandolini, Giuliano (1974). "A comparative study of the aromatic character of furan, thiophen, selenophen, and tellurophen". Journal of the Chemical Society, Perkin Transactions 2. 1974 (4): 332–337. doi:10.1039/P29740000332.
- Stein, André L.; Alves, Diego; da Rocha, Juliana T.; Nogueira, Cristina W.; Zeni, Gilson (2008). "Copper Iodide-Catalyzed Cyclization of (Z)-Chalcogenoenynes". Organic Letters. 10 (21): 4983–4986. doi:10.1021/ol802060f. ISSN 1523-7060. PMID 18826235.
- Garrett, Graham E.; Carrera, Elisa I.; Seferos, Dwight S.; Taylor, Mark S. (2016). "Anion recognition by a bidentate chalcogen bond donor". Chemical Communications. 52 (64): 9881–9884. doi:10.1039/c6cc04818h. ISSN 1359-7345. PMID 27376877.
- Karapala, Vamsi Krishna; Shih, Hong-Pin; Han, Chien-Chung (2018). "Cascade and Effective Syntheses of Functionalized Tellurophenes". Organic Letters. 20 (6): 1550–1554. doi:10.1021/acs.orglett.8b00279. ISSN 1523-7060. PMID 29494165.
- Jahnke, Ashlee A.; Djukic, Brandon; McCormick, Theresa M.; Buchaca Domingo, Ester; Hellmann, Christoph; Lee, Yunjeong; Seferos, Dwight S. (2013). "Poly(3-alkyltellurophene)s Are Solution-Processable Polyheterocycles". Journal of the American Chemical Society. 135 (3): 951–954. doi:10.1021/ja309404j. PMID 23286232.
- Becke, Axel D. (April 1993). "Density‐functional thermochemistry. III. The role of exact exchange". The Journal of Chemical Physics. 98 (7): 5648–5652. Bibcode:1993JChPh..98.5648B. doi:10.1063/1.464913. ISSN 0021-9606.
- Hay, P. Jeffrey; Wadt, Willard R. (January 1985). "Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg". The Journal of Chemical Physics. 82 (1): 270–283. Bibcode:1985JChPh..82..270H. doi:10.1063/1.448799. ISSN 0021-9606.
- Hanwell, Marcus D; Curtis, Donald E; Lonie, David C; Vandermeersch, Tim; Zurek, Eva; Hutchison, Geoffrey R (2012). "Avogadro: an advanced semantic chemical editor, visualization, and analysis platform". Journal of Cheminformatics. 4 (1): 17. doi:10.1186/1758-2946-4-17. ISSN 1758-2946. PMC 3542060. PMID 22889332.
- Schmidt, Michael W.; Baldridge, Kim K.; Boatz, Jerry A.; Elbert, Steven T.; Gordon, Mark S.; Jensen, Jan H.; Koseki, Shiro; Matsunaga, Nikita; Nguyen, Kiet A. (November 1993). "General atomic and molecular electronic structure system". Journal of Computational Chemistry. 14 (11): 1347–1363. doi:10.1002/jcc.540141112. ISSN 0192-8651. S2CID 3358041.
- Gordon, Mark S.; Schmidt, Michael W. (2005), "Advances in electronic structure theory", Theory and Applications of Computational Chemistry, Elsevier, pp. 1167–1189, doi:10.1016/b978-044451719-7/50084-6, ISBN 9780444517197
- Carrera, Elisa I.; McCormick, Theresa M.; Kapp, Marius J.; Lough, Alan J.; Seferos, Dwight S. (2013-11-19). "Thermal and Photoreductive Elimination from the Tellurium Center of π-Conjugated Tellurophenes". Inorganic Chemistry. 52 (23): 13779–13790. doi:10.1021/ic402485d. ISSN 0020-1669. PMID 24251356.
- Carrera, Elisa I.; Seferos, Dwight S. (2015). "Efficient halogen photoelimination from dibromo, dichloro and difluoro tellurophenes". Dalton Transactions. 44 (5): 2092–2096. doi:10.1039/c4dt01751j. ISSN 1477-9226. PMID 25154588.
- McCormick, Theresa M.; Carrera, Elisa I.; Schon, Tyler B.; Seferos, Dwight S. (2013). "Reversible oxidation of a water-soluble tellurophene". Chemical Communications. 49 (95): 11182–4. doi:10.1039/c3cc47338d. ISSN 1359-7345. PMID 24149322.
- Carrera, Elisa I.; Seferos, Dwight S. (2017-05-10). "Ring Opening of π-Delocalized 2,5-Diphenyltellurophene by Chemical or Self-Sensitized Aerobic Photooxidation". Organometallics. 36 (14): 2612–2621. doi:10.1021/acs.organomet.7b00240. ISSN 0276-7333.
- Nakayama, Juzo; Matsui, Tomoki; Sato, Noriko (June 1995). "Oxidation of Tetraarylselenophenes and Benzo[b]selenophene with m-Chloroperbenzoic Acid". Chemistry Letters. 24 (6): 485–486. doi:10.1246/cl.1995.485. ISSN 0366-7022.
- Jahnke, Ashlee A.; Seferos, Dwight S. (2011-04-29). "Polytellurophenes". Macromolecular Rapid Communications. 32 (13): 943–951. doi:10.1002/marc.201100151. ISSN 1022-1336. PMID 21538646.
- He, Gang; Torres Delgado, William; Schatz, Devon J.; Merten, Christian; Mohammadpour, Arash; Mayr, Lorenz; Ferguson, Michael J.; McDonald, Robert; Brown, Alex (2014-03-25). "Coaxing Solid-State Phosphorescence from Tellurophenes". Angewandte Chemie International Edition. 53 (18): 4587–4591. doi:10.1002/anie.201307373. ISSN 1433-7851. PMID 24668889.
- Rivard, Eric (2015-06-05). "Tellurophenes and Their Emergence as Building Blocks for Polymeric and Light-emitting Materials". Chemistry Letters. 44 (6): 730–736. doi:10.1246/cl.150119. ISSN 0366-7022.
- Nagahora, Noriyoshi; Yahata, Shuhei; Goto, Shoko; Shioji, Kosei; Okuma, Kentaro (2018-02-02). "2,5-Diaryltellurophenes: Effect of Electron-Donating and Electron-Withdrawing Groups on their Optoelectronic Properties". The Journal of Organic Chemistry. 83 (4): 1969–1975. doi:10.1021/acs.joc.7b02906. ISSN 0022-3263. PMID 29392944.
- Ye, Shuyang; Steube, Marvin; Carrera, Elisa I.; Seferos, Dwight S. (2016-02-12). "What Limits the Molecular Weight and Controlled Synthesis of Poly(3-alkyltellurophene)s?". Macromolecules. 49 (5): 1704–1711. Bibcode:2016MaMol..49.1704Y. doi:10.1021/acs.macromol.5b02770. ISSN 0024-9297.
- Parke, Sarah M.; Boone, Michael P.; Rivard, Eric (2016). "Marriage of heavy main group elements with π-conjugated materials for optoelectronic applications". Chemical Communications. 52 (61): 9485–9505. doi:10.1039/c6cc04023c. ISSN 1359-7345. PMID 27344980.
- Yokozawa, Tsutomu; Yokoyama, Akihiro (2009-11-11). "Chain-Growth Condensation Polymerization for the Synthesis of Well-Defined Condensation Polymers and π-Conjugated Polymers". Chemical Reviews. 109 (11): 5595–5619. doi:10.1021/cr900041c. ISSN 0009-2665. PMID 19757808.
- Al-Hashimi, Mohammed; Han, Yang; Smith, Jeremy; Bazzi, Hassan S.; Alqaradawi, Siham Yousuf A.; Watkins, Scott E.; Anthopoulos, Thomas D.; Heeney, Martin (2016). "Influence of the heteroatom on the optoelectronic properties and transistor performance of soluble thiophene-, selenophene- and tellurophene–vinylene copolymers". Chemical Science. 7 (2): 1093–1099. doi:10.1039/c5sc03501e. ISSN 2041-6520. PMC 5954972. PMID 29896373.
- Tsao, Fu An; Stephan, Douglas W. (2015). "1,1-Carboboration to tellurium–boron intramolecular frustrated Lewis pairs". Dalton Transactions. 44 (1): 71–74. doi:10.1039/c4dt03241a. ISSN 1477-9226. PMID 25408099.
- Welch, Gregory C.; Juan, Ronan R. San; Masuda, Jason D.; Stephan, Douglas W. (2006-11-17). "Reversible, Metal-Free Hydrogen Activation". Science. 314 (5802): 1124–1126. Bibcode:2006Sci...314.1124W. doi:10.1126/science.1134230. ISSN 0036-8075. PMID 17110572. S2CID 20333088.
- Tsao, Fu An; Cao, Levy; Grimme, Stefan; Stephan, Douglas W. (2015-10-12). "Double FLP-Alkyne Exchange Reactions: A Facile Route to Te/B Heterocycles". Journal of the American Chemical Society. 137 (41): 13264–13267. doi:10.1021/jacs.5b09526. ISSN 0002-7863. PMID 26447492.
- Tsao, Fu An; Stephan, Douglas W. (2018). "Synthesis and reactions of 4H-1,4-telluraborine". Chemical Communications. 54 (2): 208–211. doi:10.1039/c7cc08765a. ISSN 1359-7345. PMID 29230466.
- Li, Peng-Fei; Qian, Chenxi; Lough, Alan J.; Ozin, Geoffrey A.; Seferos, Dwight S. (2016). "Permanently porous hydrogen-bonded frameworks of rod-like thiophenes, selenophenes, and tellurophenes capped with MIDA boronates". Dalton Transactions. 45 (24): 9754–9757. doi:10.1039/c5dt04960a. ISSN 1477-9226. PMID 26758802.
- Luo, Jie; Wang, Jia-Wei; Zhang, Ji-Hong; Lai, Shan; Zhong, Di-Chang (2018). "Hydrogen-bonded organic frameworks: design, structures and potential applications". CrystEngComm. 20 (39): 5884–5898. doi:10.1039/c8ce00655e. ISSN 1466-8033.