Time–temperature superposition
The time–temperature superposition principle is a concept in polymer physics and in the physics of glass-forming liquids.[1][2][3][4] This superposition principle is used to determine temperature-dependent mechanical properties of linear viscoelastic materials from known properties at a reference temperature. The elastic moduli of typical amorphous polymers increase with loading rate but decrease when the temperature is increased.[5] Curves of the instantaneous modulus as a function of time do not change shape as the temperature is changed but appear only to shift left or right. This implies that a master curve at a given temperature can be used as the reference to predict curves at various temperatures by applying a shift operation. The time-temperature superposition principle of linear viscoelasticity is based on the above observation.[6]
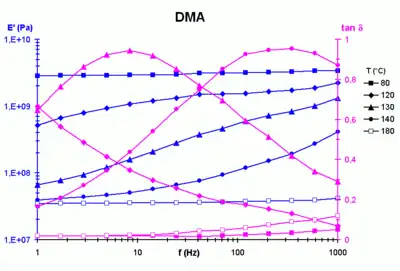
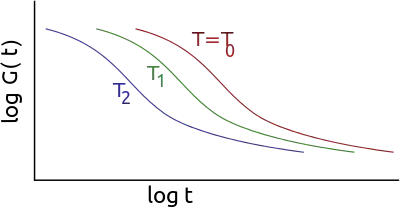
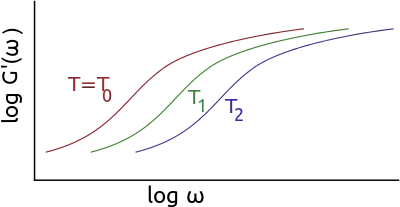
The application of the principle typically involves the following steps:
- experimental determination of frequency-dependent curves of isothermal viscoelastic mechanical properties at several temperatures and for a small range of frequencies
- computation of a translation factor to correlate these properties for the temperature and frequency range
- experimental determination of a master curve showing the effect of frequency for a wide range of frequencies
- application of the translation factor to determine temperature-dependent moduli over the whole range of frequencies in the master curve.
The translation factor is often computed using an empirical relation first established by Malcolm L. Williams, Robert F. Landel and John D. Ferry (also called the Williams-Landel-Ferry or WLF model). An alternative model suggested by Arrhenius is also used. The WLF model is related to macroscopic motion of the bulk material, while the Arrhenius model considers local motion of polymer chains.
Some materials, polymers in particular, show a strong dependence of viscoelastic properties on the temperature at which they are measured. If you plot the elastic modulus of a noncrystallizing crosslinked polymer against the temperature at which you measured it, you will get a curve which can be divided up into distinct regions of physical behavior. At very low temperatures, the polymer will behave like a glass and exhibit a high modulus. As you increase the temperature, the polymer will undergo a transition from a hard “glassy” state to a soft “rubbery” state in which the modulus can be several orders of magnitude lower than it was in the glassy state. The transition from glassy to rubbery behavior is continuous and the transition zone is often referred to as the leathery zone. The onset temperature of the transition zone, moving from glassy to rubbery, is known as the glass transition temperature, or Tg.
In the 1940s Andrews and Tobolsky[7] showed that there was a simple relationship between temperature and time for the mechanical response of a polymer. Modulus measurements are made by stretching or compressing a sample at a prescribed rate of deformation. For polymers, changing the rate of deformation will cause the curve described above to be shifted along the temperature axis. Increasing the rate of deformation will shift the curve to higher temperatures so that the transition from a glassy to a rubbery state will happen at higher temperatures.
It has been shown experimentally that the elastic modulus (E) of a polymer is influenced by the load and the response time. Time–temperature superposition implies that the response time function of the elastic modulus at a certain temperature resembles the shape of the same functions of adjacent temperatures. Curves of E vs. log(response time) at one temperature can be shifted to overlap with adjacent curves, as long as the data sets did not suffer from ageing effects[8] during the test time (see Williams-Landel-Ferry equation).
The Deborah number is closely related to the concept of time-temperature superposition.
Physical principle
Consider a viscoelastic body that is subjected to dynamic loading. If the excitation frequency is low enough[9] the viscous behavior is paramount and all polymer chains have the time to respond to the applied load within a time period. In contrast, at higher frequencies, the chains do not have the time to fully respond and the resulting artificial viscosity results in an increase in the macroscopic modulus. Moreover, at constant frequency, an increase in temperature results in a reduction of the modulus due to an increase in free volume and chain movement.
Time–temperature superposition is a procedure that has become important in the field of polymers to observe the dependence upon temperature on the change of viscosity of a polymeric fluid. Rheology or viscosity can often be a strong indicator of the molecular structure and molecular mobility. Time–temperature superposition avoids the inefficiency of measuring a polymer's behavior over long periods of time at a specified temperature by utilizing the fact that at higher temperatures and shorter time the polymer will behave the same, provided there are no phase transitions.
Time-temperature superposition

Consider the relaxation modulus E at two temperatures T and T0 such that T > T0. At constant strain, the stress relaxes faster at the higher temperature. The principle of time-temperature superposition states that the change in temperature from T to T0 is equivalent to multiplying the time scale by a constant factor aT which is only a function of the two temperatures T and T0. In other words,

The quantity aT is called the horizontal translation factor or the shift factor and has the properties:

The superposition principle for complex dynamic moduli (G* = G' + i G'' ) at a fixed frequency ω is obtained similarly:

A decrease in temperature increases the time characteristics while frequency characteristics decrease.
Relationship between shift factor and intrinsic viscosities
For a polymer in solution or "molten" state the following relationship can be used to determine the shift factor:

where ηT0 is the viscosity (non-Newtonian) during continuous flow at temperature T0 and ηT is the viscosity at temperature T.
The time–temperature shift factor can also be described in terms of the activation energy (Ea). By plotting the shift factor aT versus the reciprocal of temperature (in K), the slope of the curve can be interpreted as Ea/k, where k is the Boltzmann constant = 8.64x10−5 eV/K and the activation energy is expressed in terms of eV.
Shift factor using the Williams-Landel-Ferry (WLF) model
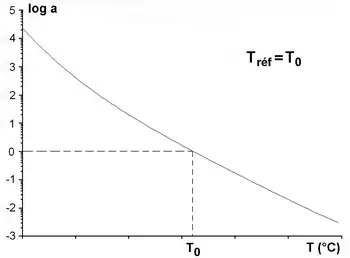
The empirical relationship of Williams-Landel-Ferry,[11] combined with the principle of time-temperature superposition, can account for variations in the intrinsic viscosity η0 of amorphous polymers as a function of temperature, for temperatures near the glass transition temperature Tg. The WLF model also expresses the change with the temperature of the shift factor.
Williams, Landel and Ferry proposed the following relationship for aT in terms of (T-T0) :

where is the decadic logarithm and C1 and C2 are positive constants that depend on the material and the reference temperature. This relationship holds only in the approximate temperature range [Tg, Tg + 100 °C]. To determine the constants, the factor aT is calculated for each component M′ and M of the complex measured modulus M*. A good correlation between the two shift factors gives the values of the coefficients C1 and C2 that characterize the material.

If T0 = Tg:

where Cg1 and Cg2 are the coefficients of the WLF model when the reference temperature is the glass transition temperature.
The coefficients C1 and C2 depend on the reference temperature. If the reference temperature is changed from T0 to T′0, the new coefficients are given by

In particular, to transform the constants from those obtained at the glass transition temperature to a reference temperature T0,

These same authors have proposed the "universal constants" Cg1 and Cg2 for a given polymer system be collected in a table. These constants are approximately the same for a large number of polymers and can be written Cg1 ≈ 15 and Cg2 ≈ 50 K. Experimentally observed values deviate from the values in the table. These orders of magnitude are useful and are a good indicator of the quality of a relationship that has been computed from experimental data.
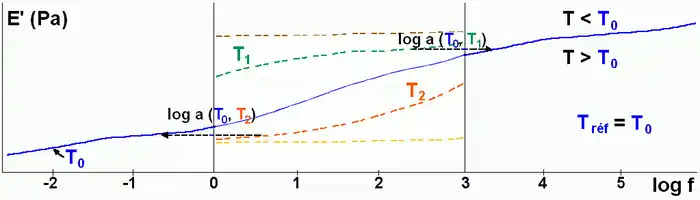
Construction of master curves
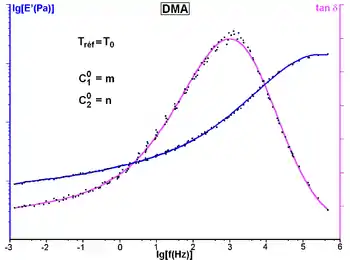
The principle of time-temperature superposition requires the assumption of thermorheologically simple behavior (all curves have the same characteristic time variation law with temperature). From an initial spectral window [ω1, ω2] and a series of isotherms in this window, we can calculate the master curves of a material which extends over a broader frequency range. An arbitrary temperature T0 is taken as a reference for setting the frequency scale (the curve at that temperature undergoes no shift).
In the frequency range [ω1, ω2], if the temperature increases from T0, the complex modulus E′(ω) decreases. This amounts to explore a part of the master curve corresponding to frequencies lower than ω1 while maintaining the temperature at T0. Conversely, lowering the temperature corresponds to the exploration of the part of the curve corresponding to high frequencies. For a reference temperature T0, shifts of the modulus curves have the amplitude log(aT). In the area of glass transition, aT is described by an homographic function of the temperature.
The viscoelastic behavior is well modeled and allows extrapolation beyond the field of experimental frequencies which typically ranges from 0.01 to 100 Hz .
Shift factor using Arrhenius law
The shift factor (which depends on the nature of the transition) can be defined, below Tg, using an Arrhenius law:

where Ea is the activation energy, R is the universal gas constant, and T0 is a reference temperature in kelvins. This Arrhenius law, under this glass transition temperature, applies to secondary transitions (relaxation) called β-transitions.
Limitations
For the superposition principle to apply, the sample must be homogeneous, isotropic and amorphous. The material must be linear viscoelastic under the deformations of interest, i.e., the deformation must be expressed as a linear function of the stress by applying very small strains, e.g. 0.01%.
To apply the WLF relationship, such a sample should be sought in the approximate temperature range [Tg, Tg + 100 °C], where α-transitions are observed (relaxation). The study to determine aT and the coefficients C1 and C2 requires extensive dynamic testing at a number of scanning frequencies and temperature, which represents at least a hundred measurement points.
References
- Hiemenz, Paul C.; Lodge, Timothy P. (2007). Polymer chemistry (2nd ed.). Taylor & Francis. pp. 486–491. ISBN 1574447793.
- Li, Rongzhi (February 2000). "Time-temperature superposition method for glass transition temperature of plastic materials". Materials Science and Engineering: A. 278 (1–2): 36–45. doi:10.1016/S0921-5093(99)00602-4.
- van Gurp, Marnix; Palmen, Jo (January 1998). "Time-temperature superposition for polymeric blends" (PDF). Rheology Bulletin. 67 (1): 5–8. Retrieved 7 December 2021.
- Olsen, Niels Boye; Christensen, Tage; Dyre, Jeppe C. (2001). "Time-Temperature Superposition in Viscous Liquids". Physical Review Letters. 86 (7): 1271–1274. arXiv:cond-mat/0006165. Bibcode:2001PhRvL..86.1271O. doi:10.1103/PhysRevLett.86.1271. ISSN 0031-9007. PMID 11178061.
- Experiments that determine the mechanical properties of polymers often use periodic loading. For such situations, the loading rate is related to the frequency of the applied load.
- Christensen, Richard M. (1971). Theory of viscoelasticity: an introduction. New York: Academic Press. p. 92. ISBN 0121742504.
- Andrews, R. D.; Tobolsky, A. V. (August 1951). "Elastoviscous properties of polyisobutylene. IV. Relaxation time spectrum and calculation of bulk viscosity". Journal of Polymer Science. 7 (23): 221–242. doi:10.1002/pol.1951.120070210.
- Struik, L. C. E. (1978). Physical aging in amorphous polymers and other materials. Amsterdam: Elsevier Scientific Pub. Co. ISBN 9780444416551.
- For the superposition principle to apply, the excitation frequency should be well above the characteristic time τ (also called relaxation time) which depends on the molecular weight of the polymer.
- Curve has been generated with data from a dynamic test with a double scanning frequency / temperature on a viscoelastic polymer.
- Ferry, John D. (1980). Viscoelastic properties of polymers (3d ed.). New York: Wiley. ISBN 0471048941.