Trace metal stable isotope biogeochemistry
Trace metal stable isotope biogeochemistry is the study of the distribution and relative abundances of trace metal isotopes in order to better understand the biological, geological, and chemical processes occurring in an environment. Trace metals are elements such as iron, magnesium, copper, and zinc that occur at low levels in the environment. Trace metals are critically important in biology and are involved in many processes that allow organisms to grow and generate energy. In addition, trace metals are constituents of numerous rocks and minerals, thus serving as an important component of the geosphere. Both stable and radioactive isotopes of trace metals exist, but this article focuses on those that are stable. Isotopic variations of trace metals in samples are used as isotopic fingerprints to elucidate the processes occurring in an environment and answer questions relating to biology, geochemistry, and medicine.
Isotope notation
In order to study trace metal stable isotope biogeochemistry, it is necessary to compare the relative abundances of isotopes of trace metals in a given biological, geological, or chemical pool to a standard (discussed individually for each isotope system below) and monitor how those relative abundances change as a result of various biogeochemical processes. Conventional notations used to mathematically describe isotope abundances, as exemplified here for 56Fe, include the isotope ratio (56R), fractional abundance (56F) and delta notation (δ56Fe). Furthermore, as different biogeochemical processes vary the relative abundances of the isotopes of a given trace metal, different reaction pools or substances will become enriched or depleted in specific isotopes. This partial separation of isotopes between different pools is termed isotope fractionation, and is mathematically described by fractionation factors α or ε (which express the difference in isotope ratio between two pools), or by "cap delta" (Δ; the difference between two δ values). For a more complete description of these notations, see the isotope notation section in Hydrogen isotope biogeochemistry.
Naturally occurring trace metal isotope variations and fractionations
In nature, variations in isotopic ratios of trace metals on the order of a few tenths to several ‰ are observed within and across diverse environments spanning the geosphere, hydrosphere and biosphere. A complete understanding of all processes that fractionate trace metal isotopes is presently lacking, but in general, isotopes of trace metals are fractionated during various chemical and biological processes due to kinetic and equilibrium isotope effects.
Geochemical fractionations
Certain isotopes of trace metals are preferentially oxidized or reduced; thus, transitions between redox species of the metal ions (e.g., Fe2+ → Fe3+) are fractionating, resulting in different isotopic compositions between the different redox pools in the environment.[1] Additionally, at high temperatures, metals ions can evaporate (and subsequently condense upon cooling), and the relative differences in isotope masses of a given heavy metal leads to fractionation during these evaporation and condensation processes.[2] Diffusion of isotopes through a solution or material can also result in fractionations, as the lighter mass isotopes are able to diffuse at a faster rate.[3] Additionally, isotopes can have slight variations in their solubility and other chemical and physical properties, which can also drive fractionation.
Biological fractionations
In sediments, oceans, and rivers, distinct trace metal isotope ratios exist due to biological processes such as metal ion uptake and abiotic processes such as adsorption to particulate matter that preferentially remove certain isotopes.[4] The trace metal isotopic composition of a given organism results from a combination of the isotopic compositions of source material (i.e., food and water) and any fractionations imparted during metal ion uptake, translocation and processing inside cells.
Applications of trace metal isotope ratios
Stable isotope ratios of trace metals can be used to answer a variety of questions spanning diverse fields, including oceanography, geochemistry, biology, medicine, anthropology and astronomy. In addition to their modern applications, trace metal isotopic compositions can provide insight into ancient biogeochemical processes operated on Earth.[5] These signatures arise because the processes that form and modify samples are recorded in the trace metal isotopic compositions of the samples. By analyzing and understanding trace metal isotopic compositions in biological, chemical or geological materials, one can answer questions such as the sources of nutrients for phytoplankton in the ocean, processes that drove the formation of geologic structures, the diets of modern or ancient organisms, and accretionary processes that took place in the early Solar System.[6][7][8] Trace metal stable isotope biogeochemistry is still an emerging field, yet each trace metal isotope system has clear, powerful applications to diverse and important questions. Important heavy metal isotope systems are discussed (in order of increasing atomic mass) in the proceeding sections.
Iron
Stable isotopes and natural abundances
Naturally occurring iron has four stable isotopes, 54Fe, 56Fe, 57Fe, and 58Fe.
| Isotope | Abundance (%) |
|---|---|
| 54Fe | 5.845 |
| 56Fe | 91.754 |
| 57Fe | 2.1191 |
| 58Fe | 0.2819 |
Stable iron isotopes are described as the relative abundance of each of the stable isotopes with respect to 54Fe. The standard for iron is elemental iron, IRMM-014, and it is distributed by the Institute for Reference Materials and Measurement. The delta value is compared to this standard, and is defined as:

Delta values are often reported as per mil values (‰), or part-per-thousand differences from the standard. Iron isotopic fractionation is also commonly described in units of per mil per atomic mass unit.
In many cases, the δ56Fe value can be related to the δ57Fe and δ58Fe values through mass-dependent fractionation:[9]


Chemistry
One of the most prevalent features of iron chemistry is its redox chemistry. Iron has three oxidation states: metallic iron (Fe0), ferrous iron (Fe2+), and ferric iron (Fe3+). Ferrous iron is the reduced form of iron, and ferric iron is the oxidized form of iron. In the presence of oxygen, ferrous iron is oxidized to ferric iron, thus ferric iron is the dominant redox state of iron at Earth's surface conditions. However, ferrous iron is the dominant redox state below the surface at depth. Because of this redox chemistry, iron can act as either an electron donor or receptor, making it a metabolically useful species.
Each form of iron has a specific distribution of electrons (i.e., electron configuration), tabulated below:
| Fe form | Electron configuration |
|---|---|
| Fe0 | [Ar]3d64s2 |
| Fe2+ | [Ar]3d6 |
| Fe3+ | [Ar]3d5 |
Equilibrium Isotope Fractionation
Variations in iron isotopes are caused by a number of chemical processes which result in the preferential incorporation of certain isotopes of iron into certain phases. Many of the chemical processes which fractionate iron are not well understood and are still being studied. The most well-documented chemical processes which fractionate iron isotopes relate to its redox chemistry, the evaporation and condensation of iron, and the diffusion of dissolved iron through systems. These processes are described in more detail below.
Fractionation as a result of redox chemistry
To first order, reduced iron favors isotopically light iron and oxidized iron favors isotopically heavy iron. This effect has been studied in regards to the abiotic oxidation of Fe2+ to Fe3+, which results in fractionation. The mineral ferrihydrite, which forms in acidic aquatic conditions, is precipitated via the oxidation of aqueous Fe2+ to Fe3+.[10] Precipitated ferrihydrite has been found to be enriched in the heavy isotopes by 0.45‰ per atomic mass unit with respect to the starting material.[10] This indicates that heavier iron isotopes are preferentially precipitated as a result of oxidizing processes.[10]
Theoretical calculations in combination with experimental data have also aimed to quantify the fractionation between Fe(III)aq and Fe(II)aq in HCl.[1] Based on modeling, the fractionation factor between the two species is temperature dependent:[1]

Fractionation as a result of evaporation and condensation
Evaporation and condensation can give rise to both kinetic and equilibrium isotope effects. While equilibrium mass fractionation is present evaporation and condensation, it is negligible compared to kinetic effects.[1] During condensation, the condensate is enriched in the light isotope, whereas in evaporation, the gas phase is enriched in the light isotope. Using the kinetic theory of gases, for 56Fe/54Fe, a fractionation factor of α = 1.01835 for the evaporation of a pool containing equimolar amounts of 56Fe and 54Fe.[1] In evaporation experiments, the evaporation of FeO at 1,823 K gave a fractionation factor of α = 1.01877.[2] Presently, there have been no experimental attempts to determine the 56Fe/54Fe fractionation factor of condensation.[1]
Fractionation as a result of diffusion
Kinetic fractionation of dissolved iron occurs as a result of diffusion. When isotopes diffuse, the lower mass isotopes diffuse more quickly than the heavier isotopes, resulting in fractionation. This difference in diffusion rates has been approximated as:[3]

In this equation, D1 and D2 are the diffusivities of the isotopes, m1 and m2 are the masses of the isotopes, and β, which can vary between 0 and 0.5, depending on the system.[9] More work is required to fully understand fractionation as a result of diffusion, studies of diffusion of iron on metal have consistently given β values of approximately 0.25.[11] Iron diffusion between silicate melts and basaltic/rhyolitic melts have given lower β values (~0.030).[12] In aqueous environments, a β value of 0.0025 has been obtained.[13]
Fractionation as a result of phase partitioning
There may be equilibrium fractionation between coexisting minerals. This would be particularly relevant when considering the formation of planetary bodies early in the solar system. Experiments have aimed to simulate the formation of the Earth at high temperatures using a platinum-iron alloy and an analog for the silicate earth at 1,500 °C.[14] However, the observed fractionation was very small, less than 0.2‰ per atomic mass unit.[14] More experimental work is needed to fully understand this effect.
Biology
In biology, iron plays a number of roles. Iron is widespread in most living organisms and is essential for their function. In microbes, iron redox chemistry is utilized as an electron donor or receptor in microbial metabolism, allowing microbes to generate energy. In the oceans, iron is essential for the growth and survival of phytoplankton, which use iron to fix nitrogen. Iron is also important in plants, given that they need iron to transfer electrons during photosynthesis. Finally, in animals, iron plays many roles, however, its most essential function is to transport oxygen in the bloodstream throughout the body. Thus, iron undergoes many biological processes, each of which have variations in which isotopes of iron they preferentially use. While iron isotopic fractionations are observed in many organisms, they are still not well understood. Improvements in the understanding the iron isotope fractionations observed in biology will enable the development of a more complete knowledge of the enzymatic, metabolic, and other biologic pathways in different organisms. Below, the known iron isotopic variations for different classes of organisms are described.
Iron reducing bacteria
Iron reducing bacteria reduce ferric iron to ferrous iron under anaerobic conditions. One of the first studies that studied iron fractionation in iron-reducing bacteria studied the bacterium Shewanella algae.[15] S. algae was grown on a ferrihydrite substrate, and was then allowed to reduce iron.[15] The study found that S. algae preferentially reduced 54Fe over 56Fe, with a δ56/54Fe value of -1.3‰.[15]
More recent experiments have studied the bacterium Shewanella putrefaciens and its reduction of Fe(III) in goethite. These studies have found δ56/54Fe values of -1.2‰ relative to the goethite.[16] The kinetics of this fractionation were also studied in this experiment, and it was suggested that the iron isotope fractionation is likely related to the kinetics of the electron transfer step.[16]
Most studies of other iron reducing bacteria have found δ56/54Fe values of approximately -1.3‰.[17][18] At high Fe(III) reduction rates, δ56/54Fe values of -2 – -3‰ relative to the substrate have been observed.[19] The study of iron isotopes in iron reducing bacteria enable the development of an improved understanding regarding the metabolic processes operating in these organisms.
| Species | δ54/56Fe (‰) | Reference |
|---|---|---|
| Fe(II)aq - FeCO3 | 0.0 | [19] |
| Fe(II)aq - Ca0.15Fe0.85CO3 | +0.9 | [19] |
| Fe(II)aq - Fe3O4 | -1.3 | [19] |
| Fe(II)aq - Ferric oxide/hydroxide | -1.3 | [15] |
| Fe(II)aq - Fe(II)s | 0.0 | [19] |
| Fe3O4 - FeCO3 | +1.3 | [19] |
| Fe3O4 - Ca0.15Fe0.85CO3 | +2.2 | [19] |
Iron oxidizing bacteria
While most iron is oxidized as a result of interaction with atmospheric oxygen or oxygenated waters, oxidation by bacteria is an active process in anoxic environments and in oxygenated, low pH (<3) environments. Studies of the acidophilic Fe(II)-oxidizing bacterium, Acidthiobacillus ferrooxidans, have been used to determine the fractionation as a result of iron-oxidizing bacteria. In most cases, δ56/54Fe values between 2 and 3‰ were measured.[20] However, a Rayleigh trend with a fractionation factor of αFe(III)aq-Fe(II)aq ~ 1.0022 was observed, which is smaller than the fractionation factor in the abiotic control experiments (αFe(III)aq-Fe(II)aq ~ 1.0034), which has been inferred to reflect a biological isotope effect.[20] Using iron isotopes, an improvement in the understanding of the metabolic processes controlling iron oxidation and energy production in these organisms can be developed.
Photoautrophic bacteria, which oxidize Fe(II) under anaerobic conditions, have also been studied. The Thiodictyon bacteria precipitate poorly crystalline hydrous ferric oxide when they oxidize iron.[21] The precipitate was enriched in the 56Fe relative to Fe(II)aq, with a δ56/54Fe value of +1.5 ± 0.2‰.[21]
Magnetotactic bacteria
Magnetotactic bacteria are bacteria with magnetosomes that contain magnetic crystals, usually magnetite or greigite, which allow them to orient themselves with the Earth’s magnetic field lines. These bacteria mineralize magnetite via the reduction of Fe(III), usually in microaerobic or anoxic environments. In the magnetotactic bacteria that have been studied, there was no significant iron isotope fractionation observed.[22]
Phytoplankton
Iron is important for the growth of phytoplankton. In phytoplankton, iron is used for electron transfer reactions in photosynthesis in both photosystem I and photosystem II.[23] Additionally, iron is an important component of the enzyme nitrogenase, which is used to fix nitrogen.[23] In measurements at open ocean stations, phytoplankton are isotopically light, with the fractionation as a result of biological uptake measured between -0.25‰ and -0.13‰.[24] Improvement in the understanding of this fractionation will enable the more precise understanding of phytoplankton photosynthetic processes.
Animals
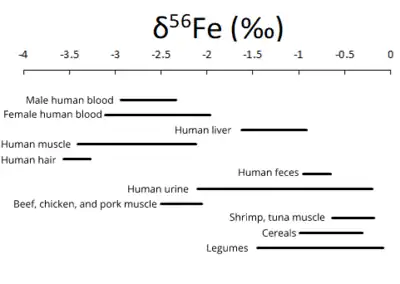
Iron has many important roles in animal biology, specifically when considering oxygen transport in the bloodstream, oxygen storage in muscles, and enzymes. Known isotope variations are shown in the figure below.[25] Iron isotopes could be useful tracers of the iron biochemical pathways in animals, and also be indicative of trophic levels in a food chain.[7]
Iron isotope variations in humans reflects a number of processes. Specifically, iron in the blood stream reflects dietary iron, which is isotopically lighter than iron in the geosphere.[26] Iron isotopes are distributed heterogeneously throughout the body, primarily to red blood cells, the liver, muscle, skin, enzymes, nails, and hair. Iron losses in the body (intestinal bleeding, bile, sweat, etc.) favor the loss of isotopically heavy iron, with mean losses averaging a δ56Fe of +10‰.[26] Iron absorption in the intestine favors lighter iron isotopes.[26] This is largely due to the fact that iron is carried by transport proteins and transferrin, both of which are kinetic processes, resulting in the preferential uptake of isotopically light iron.[26]
The observed iron isotopic variations in humans and animals are particularly important as tracers. Iron isotopic signatures are utilized to determine the geographic origin of food. Additionally, anthropologists and paleontologists use iron isotope data in order to track the transfer of iron between the geosphere and the biosphere, specifically between plant foods and animals. This allows for the reconstruction of ancient dietary habits based on the variations in iron isotopes in food.
Geochemistry
By mass, iron is the most common element on Earth, and it is the fourth most abundant element in the Earth's crust. Thus, iron is widespread throughout the geosphere, and is also common on other planetary bodies. Natural variations in the iron in the geosphere are relatively small. Currently, the values of δ56/54Fe measured in rocks and minerals range from -2.5‰ to +1.5‰. Iron isotope composition is homogeneous in igneous rocks to ±0.05‰, indicating that much of the geologic isotopic variability is a result of the formation of rocks and minerals at low temperature.[17] This homogeneity is particularly useful when tracing processes which result in fractionation through the system. While fractionation of igneous rocks is relatively constant, there are larger variations in the iron isotopic composition of chemical sediments.[27] Thus, iron isotopes are used to determine the origin of the protolith of heavily metamorphosed rocks of a sedimentary origin.[27] Improvements of the understanding regarding the way in which iron isotopes fractionate in the geosphere can help to better understand geologic processes of formation.
Natural iron isotopic variations
To date, iron is one of the most widely studied trace metals, and iron isotope compositions are relatively well-documented. Based on measurements, iron isotopes exhibit minimal variation (±3‰) in the terrestrial environment. A list of iron isotopic values of different materials from different environments is presented below.

In terrestrial environments
There is an extreme constancy of the isotopic composition of igneous rocks. The mean value of δ56Fe of terrestrial rocks is 0.00 ± 0.05‰.[17] More precise isotopic measurements indicate that the small deviations from 0.00‰ may reflect a slight mass-dependent fractionation.[17] This mass fractionation has been proposed to be FFe = 0.039 ± 0.008‰ per atomic mass unit relative to IRMM-014.[28] There may also be slight isotopic variations in igneous rocks depending on their composition and process of formation. The average value of δ56Fe for ultramafic igneous rocks is -0.06‰, whereas the average value of δ56Fe for mid-ocean ridge basalts (MORB) is +0.03‰.[17] Sedimentary rocks exhibit slightly larger variations in δ56Fe, with values between -1.6‰ and +0.9‰ relative to IRMM-014.[28] Banded iron formations δ56Fe span the entire range observed on Earth, from -2.5‰ to +1‰.[5]
In the oceans
There are slight iron isotopic variations in the oceans relative to IRMM-014, which likely reflect variations in the biogeochemical cycling of iron within a given ocean basin. In the southeastern Atlantic, δ56Fe values between -0.13 and +0.21‰ have been measured.[29] In the north Atlantic, δ56Fe values between -1.35 and +0.80‰ have been measured.[30][31] In the equatorial Pacific δ56Fe values between -0.03 and +0.58‰ have been measured.[32][33] The supply of aerosol iron particles to the ocean have an isotopic composition of approximately 0‰.[1] Dissolved iron riverine input to the ocean is isotopically light relative to igneous rocks, with δ56Fe values between -1 and 0‰.[1]
Most modern marine sediments have δ56Fe values similar to those of igneous δ56Fe values.[1] Marine ferromanganese nodules have δ56Fe values between -0.8 and 0‰.
In hydrothermal systems
Hot (> 300 °C) hydrothermal fluids from mid ocean ridges are isotopically light, with δ56Fe between -0.2 and -0.8‰.[1] Particles in hydrothermal plumes are isotopically heavy relative to the hydrothermal fluids, with δ56Fe between 0.1 and 1.1‰.[1] Hydrothermal deposits have average δ56Fe between -1.6 and 0.3‰.[1] The sulfide minerals within these deposits have δ56Fe between -2.0 and 1.1‰.[1]
In extraterrestrial objects
Variations in iron isotopic composition have been observed in meteorite samples from other planetary bodies. The Moon has variations in iron isotopes of 0.4‰ per atomic mass unit.[34] Mars has very small isotope fractionation of 0.001 ± 0.006‰ per atomic mass unit.[8] Vesta has iron fractionations of 0.010 ± 0.010‰ per atomic mass unit.[8] The chondritic reservoir exhibits fractionations of 0.069 ± 0.010‰ per atomic mass unit.[8] Isotopic variations observed on planetary bodies can help to constrain and better understand their formation and processes occurring in the early Solar System.
Measurement
High precision iron isotope measurements are obtained either via thermal ionization mass spectrometry (TIMS) or multi-collector inductively coupled plasma mass spectrometry (MC-ICP-MS).
Applications of iron isotopes
Iron isotopes have many applications in the geosciences, biology, medicine, and other fields. Their ability to act as isotopic tracers allows for their use to determine information regarding the formation of geologic units and as a potential proxy for life on Earth and other planets. Iron isotopes also have applications in anthropology and paleontology, as they are used to study the diets of ancient civilizations and animals. The widespread uses of iron in biology make its isotopes a promising frontier in biomedical research, specifically their use to prevent and treat blood conditions and other pathological blood diseases. Some of the more prevalent applications of iron isotopes are described below.
Banded iron formations
Banded iron formations (BIFs) are particularly important when considering the surface environments of the early Earth, which were significantly different from the surface environments observed today. This is manifested in the mineralogy of these formations, which are indicative of different redox conditions.[35] Additionally, BIFs are interesting in that they were deposited while major changes were occurring in the atmosphere and in the biosphere 2.8 to 1.8 billion years ago.[35] Iron isotopic studies can reveal the details of the formation of BIFs, which allows for the reconstruction of redox and climatic conditions at the time of deposition.
BIFs formed as a result of the oxidation of iron by oxygen, which was likely generated by the evolution of cyanobacteria.[36] This was followed by the subsequent precipitation of iron particles in the ocean.[36] Observed variations in the iron isotopic composition of BIFs span the entire range observed on Earth, with δ56/54Fe values between -2.5 and +1‰.[5] The cause of these variations are hypothesized to occur for three reasons. The first relates to the varying mineralogy of the BIFs. Within the BIFs, minerals such as hematite, magnetite, siderite, and pyrite are observed.[5] These minerals each having varying isotopic fractionation, likely as a result of their structures and the kinetics of their growth.[5] The isotopic composition of the BIFs is indicative of the fluids from which they precipitated, which has applications when reconstructing environmental conditions of the ancient Earth.[5] It has also been suggested that BIFs may be biologic in origin. The range of their δ56/54Fe values fall within the range of those observed to occur as a result of biologic processes relating to bacterial metabolic processes, such as those of anoxygenic phototrophic iron-oxidizing bacteria.[36] Ultimately, the improved understanding of BIFs using iron isotope fractionations would allow for the reconstruction of past environments and the constraint of processes occurring on the ancient Earth. However, given that the values observed as a result of biogenic and abiogenic fractionation are relatively similar, the exact processes of BIFs are still unclear. Thus, the continued study and improved understanding of biologic and abiologic fractionation effects would be beneficial in providing better details regarding BIF formation.
Iron cycling in the ocean

Iron isotopes have become particularly useful in recent years for tracing biogeochemical cycling in the oceans. Iron is an important micronutrient for living species in the ocean, particularly for the growth of phytoplankton. Iron is estimated to limit phytoplankton growth in about one half of the ocean.[38] As a result, the development of a better understanding of sources and cycling of iron in the modern oceans is important. Iron isotopes have been used to better constrain these pathways through data collected by the GEOTRACES program, which has collected iron isotopic data throughout the ocean. Based on the variations in iron isotopes, biogeochemical cycling and other processes controlling iron distribution in the ocean can be elucidated.
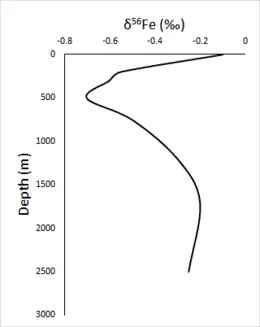
For example, the combination of iron concentration and iron isotope data can use to determine the sources of oceanic iron. In the South Atlantic and in the Southern Ocean, isotopically light iron is observed in intermediate waters (200 - 1,300 meters), whereas isotopically heavy iron is observed in surface waters and deep waters (> 1,300 meters).[38] To first order, this demonstrates that there are different sources, sinks, and processes contributing to the iron cycle in varying water masses. The isotopically light iron in intermediate waters suggests that the dominant iron sources include remineralized organic matter.[38] This organic matter is isotopically light because phytoplankton preferentially take up light iron.[38] In the surface ocean, the isotopically heavy iron represents the external sources of iron, such as dust, which is isotopically heavy relative to IRMM-014, and the sink of light isotopes as a result of their preferential uptake by phytoplankton.[38] The isotopically heavy iron in the deep ocean suggests that the iron cycle is dominated by the abiotic, non-reductive release of iron, via desorption or dissolution, from particles.[38] Isotopic analyses similar to the one above are utilized throughout all of the world's oceans to better understand regional variability in the processes which control iron cycling.[39][40] These analyses can then be synthesized to better model the global biogeochemical cycling of iron, which is particularly important when considering primary production in the ocean.
Constraining processes on extraterrestrial bodies
Iron isotopes have been applied for a number of purposes on planetary bodies. Their variations have been measured to more precisely determine the processes that occurred during planetary accretion. In the future, the comparison of observed biological fractionation of iron on Earth to fractionation on other planetary bodies may have astrobiological implications.
Planetary accretion
One of the primary challenges in the study of planetary accretion is the fact that many tracers of the processes occurring in the early Solar System have been eliminated as a result of subsequent geologic events. Because transition metals do not show large stable isotope fractionations as a result of these events and because iron is one of the most abundant elements in the terrestrial planets, its isotopic variability has been used as a tracer of early Solar System processes.[41][8]
| Planetary body | δ57/54Fe |
|---|---|
| Vesta | 0.031 |
| Mars | 0.03 |
| Moon | 0.029 |
| Mafic Earth | 0.032 |
Variations in δ57/54Fe between samples from Vesta, Mars, the Moon, and Earth have been observed, and these variations cannot be explained by any known petrological, geochemical, or planetary processes, thus, it has been inferred that the observed fractionations are a result of planetary accretion.[8] It is interesting to note that the isotopic compositions of the Earth and the Moon are much heavier than that of Vesta and Mars. This provides strong support for the giant-impact hypothesis as an impact of this energy would generate large amounts of energy, which would melt and vaporize iron, leading to the preferential escape of the lighter iron isotopes to space.[8] More of the heavier isotopes would remain, resulting in the heavier iron isotopic compositions observed for the Earth and the Moon. The samples from Vesta and Mars exhibit minimal fractionation, consistent with the theory of runaway growth for their formations, as this process would not yield significant fractionations.[8] Further study of the stable isotope of iron in other planetary bodies and samples could provide further evidence and more precise constraints for planetary accretion and other processes that occurred in the early Solar System.
Astrobiology
The use of iron isotopes may also have applications when studying potential evidence for life on other planets. The ability of microbes to utilize iron in their metabolisms makes it possible for organisms to survive in anoxic, iron-rich environments, such as Mars. Thus, the continual improvement of knowledge regarding the biological fractionations of iron observed on Earth can have applications when studying extraterrestrial samples in the future.[42] While this field of research is still developing, this could provide evidence regarding whether a sample was generated as a result of biologic or abiologic processes depending on the isotopic fractionation. For example, it has been hypothesized that magnetite crystals found in Martian meteorites may have formed biologically as a result of their striking similarity to magnetite crystals produced by magnetotactic bacteria on Earth.[43] Iron isotopes could be used to study the origin of the proposed "magnetofossils" and other rock formations on Mars.[43]
Biomedical research
Iron plays many roles in human biology, specifically in oxygen transport, short-term oxygen storage, and metabolism[44] Iron also plays a role in the body's immune system.[4] Current biomedical research aims to use iron isotopes to better understand the speciation of iron in the body, with hopes of eventually being able to reduce the availability of free iron, as this would help to defend against infection.[4]
Iron isotopes can also be utilized to better understand iron absorption in humans.[26] The iron isotopic composition of blood reflects an individual's long-term absorption of dietary iron.[26] This allows for the study of genetic predisposition to blood conditions, such as anemia, which will ultimately enable the prevention, identification, and resolution of blood disorders.[26] Iron isotopic data could also aid in identifying impairments of the iron absorption regulatory system in the body, which would help to prevent the development of pathological conditions related to issues with iron regulation.[45]
Cobalt
Copper
Stable isotopes and natural abundances
Copper has two naturally occurring stable isotopes: 63Cu and 65Cu, which exist in the following natural abundances:
| Isotope | Abundance (%) |
|---|---|
| 63Cu | 69.17 |
| 65Cu | 30.83 |
The isotopic composition of Cu is conventionally reported in delta notation (in ‰) relative to a NIST SRM 976 standard:
![{\displaystyle \delta ^{65}Cu=\left[{\frac {(^{65}Cu/^{63}Cu)_{sample}}{(^{65}Cu/^{63}Cu)_{NIST976}}}\right]}](../I/2460241f51ef018d937f8f6751d60b9d239cc424.svg)
Chemistry
Copper can exist in non-ionic form (as Cu0) or in one of two redox states: Cu1+ (reduced) or Cu2+ (oxidized). Each form of Cu has a specific distribution of electrons (i.e., electron configuration), tabulated below:
| Cu form | Electron configuration |
|---|---|
| Cu0 | [Ar] 3d10 4s1 |
| Cu1+ | [Ar] 3d10 4s0 |
| Cu2+ | [Ar] 3d9 4s0 |
The electronic configurations of Cu control the number and types of bonds Cu can form with other atoms (e.g., see Copper Biology section). These diverse coordination chemistries are what enable Cu to participate in many different biological and chemical reactions.
Finally, due to its full d-orbital, Cu1+ has diamagnetic resonance. In contrast, Cu2+ has one unpaired electron in its d-orbital, giving it paramagnetic resonance. The different resonances of the Cu ions enable determination of Cu's redox state by techniques such as electron paramagnetic resonance (epr) spectroscopy, which can identify atoms with unpaired electrons by exciting electron spins.
Equilibrium isotope fractionation
Transitions between redox species Cu1+ and Cu2+ fractionate Cu isotopes. 63Cu2+ is preferentially reduced over 65Cu2+, leaving the residual Cu2+ enriched in 65Cu. The equilibrium fractionation factor for speciation between Cu2+ and Cu1+ (αCu(II)-Cu(I)) is 1.00403 (i.e., dissolved Cu2+ is enriched in 65Cu by ~+4‰ relative to Cu1+).[46][47]
Biology
Copper can be found in the active sites of most enzymes that catalyze redox reactions (i.e., oxidoreductases), as it facilitates single electron transfers while reversibly oscillating between the Cu1+ and Cu2+ redox states. Enzymes typically contain between one (mononuclear) and four (tetranuclear) copper centers, which enable enzymes to catalyze different reactions. These copper centers coordinate with different ligands depending on the Cu redox state. Oxidized Cu2+ preferentially coordinates with "hard donor" ligands (e.g., N- or O-containing ligands such as histidine, aspartic acid, glutamic acid or tyrosine), while reduced Cu1+ preferentially coordinates with "soft donor" ligands (e.g., S-containing ligands such as cysteine or methionine).[48] Copper's powerful redox capability makes it critically important for biology, but comes at a cost: Cu1+ is a highly toxic metal to cells because it readily abstracts single electrons from organic compounds and cellular material, leading to production of free radicals. Thus, cells have evolved specific strategies for carefully controlling the activity of Cu1+ while exploiting its redox behavior.
Examples of copper-based enzymes
Copper serves catalytic and structural roles in many essential enzymes in biology. In the context of catalytic activity, copper proteins function as electron or oxygen carriers, oxidases, mono- and dioxygenases and nitrite reductases. In particular, copper-containing enzymes include hemocyanins, one flavor of superoxide dismutase (SOD), metallothionein, cytochrome c oxidase, multicopper oxidase and particulate methane monooxygenase (pMMO).
Biological fractionation
Biological processes that fractionate Cu isotopes are not well-understood, but play an important role in driving the δ65Cu values of materials observed in the marine and terrestrial environments. The natural 65Cu/63Cu varies according to copper's redox form and the ligand to which copper binds. Oxidized Cu2+ preferentially coordinates with hard donor ligands (e.g., N- or O-containing ligands), while reduced Cu1+ preferentially coordinates with soft donor ligands (e.g., S-containing ligands).[48] As 65Cu is preferentially oxidized over 63Cu, these isotopes tend to coordinate with hard and soft donor ligands, respectively.[48] Cu isotopes can fractionate upon Cu-bacteria interactions from processes that include Cu adsorption to cells, intracellular uptake, metabolic regulation and redox speciation.[46] Fractionation of Cu isotopes upon adsorption to cellular walls appears to depend on the surface functional groups that Cu complexes with, and can span positive and negative values. Furthermore, bacteria preferentially incorporate the lighter Cu isotope intracellularly and into proteins. For example, E. coli, B. subtilis and a natural consortia of microbes sequestered Cu with apparent fractionations (ε65Cu) ranging from ~-1.0 to -4.4‰. Additionally, fractionation of Cu upon incorporation into the apoprotein of azurin was ~-1‰ in P. aeruginosa, and -1.5‰ in E. coli, while ε65Cu values of Cu incorporation into Cu-metallothionein and Cu-Zn-SOD in yeast were -1.7 and -1.2‰, respectively.[46]
Geochemistry
The concentration of Cu in bulk silicate Earth is ~30 ppm,[50] slightly less than its average concentration (~72 ppm) in fresh mid-oceanic ridge basalt (MORB) glass.[51] Cu1+ and Cu2+ form a variety of sulfides (often in association with Fe), as well as carbonates and hydroxides (e.g., chalcopyrite, chalcocite, cuprite and malachite). In mafic and ultramafic rocks, Cu tends to be concentrated in sulfidic materials.[51] In freshwater, the predominant form of Cu is free Cu2+; in seawater, Cu complexes with carbonate ligands to form CuCO3 and [Cu(CO3)2]2−.
Measurement
In order to measure Cu isotope ratios of various materials, several steps must be taken prior to the isotopic measurement in order to extract and purify copper. The first step in the analytical pipeline to measure Cu isotopes is to liberate Cu from its host material. Liberation should be quantitative, otherwise fractionation may be introduced at this step. Cu-containing rocks are generally dissolved with HF; biological materials are commonly digested with HNO3. Seawater samples must be concentrated due to the low (nM) concentrations of Cu in the ocean. The sample material is subsequently run through an anion-exchange column to isolated and purify Cu. This step can also introduce Cu isotope fractionation if Cu is not quantitatively recovered from the column.[52] If samples are from seawater, other ions (e.g., Na+, Mg2+, Ca2+) must be removed in order to eliminate isobaric interferences during the isotope measurement. Prior to 1992, 65Cu/63Cu ratios were measured via thermal ionization mass spectrometry (TIMS).[53][54] Today, Cu isotopic compositions are measured via multi-conductor inductively coupled plasma mass spectrometry (MC-ICP-MS), which ionizes samples using inductively coupled plasma and introduces smaller errors than TIMS.
Natural copper isotopic variations
The field of Cu isotope biogeochemistry is still in a relatively early stage, so the Cu isotope compositions of materials in the environment are not well-documented. However, based on a compilation of measurements already made, it appears that Cu isotope ratios vary somewhat widely within and between environmental materials (e.g., plants, minerals, seawater, etc.), though as a whole, these ratios do not vary by more than ±10‰.

In humans
In human bodies, coppers is an important constituent of many essential enzymes, including ceruloplasmin (which carries Cu and oxidizes Fe2+ in human plasma), cytochrome c oxidase, metallothionein and superoxide dismutase 1.[55] Serum in human blood is typically 65Cu-depleted by ~0.8‰ relative to erythrocytes (i.e., red blood cells). In a study of 49 male and female blood donors, the average δ65Cu value of the donors' blood serum was -0.26 ± 0.40‰, while that of their erythrocytes was +0.56 ± 0.50‰.[56] In a separate study, δ65Cu values of serum in 20 healthy patients ranged from -0.39 to +0.38‰, while the δ65Cu values of their erythrocytes ranged from +0.57 to +1.24‰.[57] To balance Cu loss due to menstruation, a large portion of Cu in the blood of menstruating women comes from their liver. Due to fractionation associated with Cu transport from the liver to the blood, the total blood of pre-menopausal women is generally 65Cu-depleted relative to that of males and non-menstruating women.[58] The δ65Cu values of healthy human liver tissue in 7 patients ranged from -0.45 to -0.11‰.[57]
In the terrestrial environment
To first order, δ65Cu values in organisms are driven by the δ65Cu values of source materials. The δ65Cu values of various soils from different regions have been found to vary from -0.34 to +0.33‰[59][60] depending on the biogeochemical processes taking place in the soil and the ligands with which Cu complexes. Organic-rich soils generally have lighter δ65Cu values than mineral soils because the organic layers result from plant litter, which is isotopically light.[59]
In plants, δ65Cu values vary between the different components (seeds, roots, stem and leaves). The δ65Cu values the roots of rice, lettuce, tomato and durum wheat plants were found to be 0.5 to 1.0‰ 65Cu-depleted relative to their source, while their shoots were up to 0.5‰ lighter than the roots.[61][62][63] Seeds appear to be the most isotopically light component of plants, followed by leaves, then stems.[63]
Rivers sampled throughout the world have a range of dissolved δ65Cu values from +0.02 to +1.45‰.[64] The average δ65Cu values of the Amazon, Brahmaputra and Nile rivers are 0.69, 0.64 and 0.58‰, respectively. The average δ65Cu value of the Chang Jiang river is 1.32‰, while that of the Missouri river is 0.13‰.[64]


In rocks and minerals
In general, igneous, metamorphic and sedimentary processes do not appear to strongly fractionate Cu isotopes, while δ65Cu values of Cu minerals vary widely. The average Cu isotopic composition of bulk silicate Earth has been measured as 0.06 ± 0.20‰ based on 132 different terrestrial samples.[67] MORBs and oceanic island basalts (OIBs) generally have homogenous Cu isotopic compositions that fall around 0‰,[67][68][69][70] while arc and continental basalts have more heterogeneous Cu isotope compositions that range from -0.19 to +0.47‰.[67] These Cu isotope ratios of basalts suggest that mantle partial melting imparts negligible Cu isotopic fractionation, while recycling of crustal materials leads to widely variable δ65Cu values.[67] The Cu isotope compositions of copper-containing minerals vary over a wide range, likely due to alteration of the primary high-temperature deposits.[51] In one study that investigated Cu isotopic compositions of various minerals from hydrothermal fields along the mid-Atlantic ridge, chalcopyrite from mafic igneous rocks had δ65Cu values of -0.1 to -0.2‰, while Cu minerals in black smokers (chalcopyrite, bornite, covellite and atacamite) exhibited a wider range of δ65Cu values from -1.0 to +4.0‰.[71] Additionally, atacamite lining the outer rims of black smokers can be up to 2.5‰ heavier than chalcopyrite contained within the black smoker. δ65Cu values of Cu minerals (including chrysocolle, azurite, malachite, cuprite and native copper) in low-temperature deposits have been observed to vary widely over a range of -3.0 to +5.6‰.[72][71]
In the marine environment
Cu is strongly cycled in the surface and deep ocean. In the deep ocean, Cu concentrations are ~5 nM in the Pacific[65] and ~1.5 nM in the Atlantic.[66] The deep/surface ratio of Cu in the ocean is typically <10, and vertical concentration profiles for Cu are roughly linear due to biological recycling and scavenging processes[6] as well as adsorption to particles.
Due to equilibrium and biological processes that fractionate Cu isotopes in the marine environment, the bulk copper isotopic composition (δ65Cu = +0.6 to +1.5‰) is different from the δ65Cu values of the riverine input (δ65Cu = +0.02 to +1.45‰, with discharge-weighted average δ65Cu = +0.68‰) to the oceans.[64][66][73] δ65Cu values of the surface layers of FeMn-nodules are fairly homogenous throughout the oceans (average = 0.31‰),[69] suggesting low biological demand for Cu in the marine environment compared to that of Fe or Zn. Additionally, δ65Cu values in the Atlantic ocean do not markedly vary with depth, ranging from +0.56 to +0.72‰.[66] However, Cu isotope compositions of material collected on sediment traps at depths of 1,000 and 2,500 m in the central Atlantic ocean show seasonal variation with heaviest δ65Cu values in the spring and summer seasons[68] suggesting seasonal preferential uptake of 63Cu by biological processes.
Equilibrium processes that fractionate Cu isotopes include high temperature ion exchange and redox speciation between mineral phases, and low temperature ion exchange between aqueous species or redox speciation between inorganic species.[46] In riverine and marine environments, 65Cu/63Cu ratios are driven by preferential adsorption of 63Cu to particulate matter and preferential binding of 65Cu to organic complexes.[64] As a net result, ocean sediments tend to be depleted in 63Cu relative to the bulk ocean. For example, the downcore δ65Cu values of a 760 cm sedimentary core taken from the Central Pacific ocean varied from -0.94 to -2.83‰, significantly lighter than the bulk ocean.[51]
Medicine
Due to its relatively short turnover time of ~6 weeks in the human body,[74] Cu serves as an important indicator of cancer and other diseases that rapidly evolve. The serum of cancer patients contains significantly higher levels of Cu than that of healthy patients due to copper chelation by lactate, which is produced via anaerobic glycolysis by tumor cells.[55] These imbalances in Cu homeostasis are reflected isotopically in the serum and organ tissues of patients with various types of cancer, where the serum of cancer patients is generally 65Cu-depleted relative to the serum of healthy patients, while organ tumors are generally 65Cu-enriched.[75] In one study,[57] the blood components of patients with hepatocellular carcinomas (HCC) was found to be, on average, depleted in 65Cu by 0.4‰ relative to the blood of non-cancer patients. In particular, the δ65Cu values of the serum in patients with HCC ranged from -0.66 to +0.47‰ (compared to serum δ65Cu values of -0.39 to +0.38‰ in matched control patients), and the δ65Cu values of the erythrocytes in the HCC patients ranged from -0.07 to +0.92‰ (compared to erythrocyte δ65Cu values of +0.57 to +1.24‰ in matched control patients). The liver tumor tissues in the HCC patients were 65Cu-enriched relative to healthy liver tissue in the same patients (δ65Culiver, HCC = -0.02 to +0.43‰; δ65Culiver, healthy = -0.45 to -0.11‰), and the magnitude of 65Cu-enrichment mirrored that of the 65Cu-depletion observed in the cancer patients' serum. Though our understanding of how copper isotopes are fractionated during cancer physiologies is still limited, it is clear that copper isotope ratios may serve as a powerful biomarker of cancer presence and progression.
Zinc
Stable isotopes and natural abundances
Zinc has five stable isotopes, tabulated along with their natural abundances below:
| Isotope | Abundance (%) |
|---|---|
| 64Zn | 48.63 |
| 66Zn | 27.90 |
| 67Zn | 4.10 |
| 68Zn | 18.75 |
| 70Zn | 0.62 |
The isotopic composition of Zn is reported in delta notation (in ‰):
![{\displaystyle \delta ^{x}Zn=\left[{\frac {(^{x}Zn/^{64}Zn)_{sample}}{(^{x}Zn/^{64}Zn)_{std}}}\right]}](../I/8a0f7672f5164b841ea808cd3ccde6529261fa74.svg)
where xZn is a Zn isotope other than 64Zn (commonly either 66Zn or 68Zn). Standard reference materials used for Zn isotope measurements are JMC 3-0749C, NIST-SRM 683 or NIST-SRM 682.
Chemistry
Because it has just one valence state (Zn2+), zinc is a redox-inert element. The electronic configurations of Zn0 and Zn2+ are shown below:
| Zn form | Electron configuration |
|---|---|
| Zn0 | [Ar] 3d10 4s2 |
| Zn2+ | [Ar] 3d10 4s0 |
Biology
Zinc is present in almost 3,000 human proteins, and thus is essential for nearly all cellular functions.[76] Zn is also a key constituent of enzymes involved in cell regulation.[77] Consistent with its ubiquitous presence, total cellular Zn concentrations are typically very high (~200 μM),[78] while the concentrations of free Zn ions in the cytoplasms of cells can be as low as a few hundred picomolar,[79][80] maintained within a narrow range to avoid deficiency and toxicity. One feature of Zn that makes it so critical in cellular biology is its flexibility in coordination to different numbers and types of ligands. Zn can coordinate with anywhere between three and six N-, O- and S-containing ligands (such as histidine, glutamic acid, aspartic acid and cysteine), resulting in a large number of possible coordination chemistries. Zn tends to bind to metal sites of proteins with relatively high affinities compared to other metal ions which, aside from its important functions in enzymatic reactions, partly explains its ubiquitous presence in cellular enzymes.[78]
Examples of zinc-based enzymes
Zn is present in the active sites of most hydrolytic enzymes, and is used as an electrophilic catalyst to activate water molecules that ultimately hydrolyze chemical bonds. Examples of zinc-based enzymes include superoxide dismutase (SOD), metallothionein, carbonic anhydrase, Zn finger proteins, alcohol dehydrogenase and carboxypeptidase.
Biological fractionation
Relatively little is known about isotopic fractionation of zinc by biological processes, but several studies have elucidated that Zn isotopes fractionate during surface adsorption, intracellular uptake processes and speciation. Many organisms, including certain species of fish, plants and marine phytoplankton, have both high- and low-affinity Zn transport systems, which appear to fractionate Zn isotopes differently. A study by John et al.[81] observed apparent isotope effects associated with Zn uptake by the marine diatom Thalassiosira oceanica of -0.2‰ for high-affinity uptake (at low Zn concentrations) and -0.8‰ for low-affinity uptake (at high Zn concentrations). Additionally, in this study, unwashed cells were enriched in 65Zn, indicating preferential adsorption of 65Zn to the extracellular surfaces of T. oceanica. Results from John et al.[81] demonstrating apparent discrimination against the heavy isotope (66Zn) during uptake conflict with results by Gélabert et al.[82] in which marine phytoplankton and freshwater periphytic organisms preferentially uptook 66Zn from solution. The latter authors explained these results as due to a preferential partitioning of 66Zn into a tetrahedrally coordinated structure (i.e., with carboxylate, amine or silanol groups on or inside the cell) over an octahedral coordination with six water molecules in the aqueous phase, consistent with quantum mechanical predictions. Kafantaris and Borrok[83] grew model organisms B. subtilis, P. mendocina and E. coli, as well as a natural bacterial consortium collected from soil, on high and low concentrations of Zn. In the high [Zn] condition, the average fractionation of Zn isotopes imparted by cellular surface adsorption was +0.46‰ (i.e., 66Zn was preferentially adsorbed), while fractionation upon intracellular incorporation varied from -0.2 to +0.5‰ depending on the bacterial species and growth phase. Empirical models of the low [Zn] condition estimated larger Zn isotope fractionation factors for surface adsorption ranging from +2 to +3‰. Overall, Zn isotope ratios in microbes appear to be driven by a number of complex factors including surface interactions, bacterial metal metabolism and metal speciation, but by understanding the relative contributions of these factors to Zn isotope signals, one can use Zn isotopes to investigate metal-binding pathways operating in natural communities of microbes.
Geochemistry
The concentration of Zn in bulk silicate Earth is ~55 ppm,[50] while its average concentration in fresh mid-oceanic ridge basalt (MORB) glass is ~87 ppm.[51] Like Cu, Zn commonly associates with Fe to form a variety of zinc sulfide minerals such as sphalerite. Additionally, Zn associates with carbonates and hydroxides to form numerous diverse minerals (e.g., smithsonite, sweetite, etc.). In mafic and ultramafic rocks, Zn tends to concentrate in oxides such as spinel and magnetite. In freshwater, Zn predominantly complexes with water to form an octahedrally coordinated aqua ion [Zn(H2O)6]2+. In seawater, Cl− ions replace up to four water molecules in the Zn aqua ion, forming [ZnCl(H2O)5]+, [ZnCl2(H2O)4]0 and [ZnCl4(H2O)2]−.[84][85]
Measurement
The analytical pipeline for preparation of sample material for Zn isotope measurements is similar to that of Cu, consisting of digestion of host material or concentration from seawater, isolation and purification via anion-exchange chromatography, removal of ions of interfering mass (in particular, 64Ni) and isotope measurement via MC-ICP-MS (see Copper Isotope Measurement section for more details).
Natural zinc isotopic variations
As with Cu, the field of Zn isotope biogeochemistry is still in a relatively early stage, so the Zn isotope compositions of materials in the environment are not well-documented. However, based on a compilation of some reported measurements, it appears that Zn isotope ratios do not vary widely among environmental materials (e.g., plants, minerals, seawater, etc.), as δ66Zn values of materials typically fall within a range of -1 to +1‰.

In humans
Zn isotope ratios vary between individual blood components, bones and the different organs in humans, though in general, δ66Zn values fall within a narrow range. In the blood of healthy individuals, the Zn isotopic composition of erythrocytes is typically ~0.3‰ lighter than that of serum, and no significant differences in erythrocyte or serum δ66Zn values exist between men and women. For example, in the blood of 49 healthy blood donors, the average erythrocyte δ66Zn value was +0.44 ± 0.33‰, while that of serum was +0.17 ± 0.26‰.[56] In a separate study on 29 donors, a similar average δ66Zn value of +0.29 ± 0.27‰ was obtained for the patients' serum.[86] Additionally, in a small sample set of volunteers, whole blood δ66Zn values were ~+0.15‰ higher for vegetarians than for omnivores,[87] suggesting diet plays an important role in driving Zn isotope compositions in the human body.
In the terrestrial environment
Zn isotope ratios vary on small scales throughout the terrestrial biosphere. Zn is released into soils during mineral weathering, and isotopes of Zn fractionate upon interaction with mineral and organic components in the soil. In 5 soil profiles collected from Iceland (all derived from the same parent basalt), soil δ66Zn values varied from +0.10 to +0.35‰, and the organic-rich layers were 66Zn-depleted relative to the mineral-rich layers, likely due to contribution by isotopically light organic matter and Zn loss by leaching.[88]
Isotopic discrimination of Zn varies in different components of higher plants, likely due to the various processes involved in Zn uptake, binding, transport, diffusion, speciation and compartmentalization. For example, Weiss et al.[61] observed heavier δ66Zn values in the roots of several plants (rice, lettuce and tomato) relative to the bulk solution in which the plants were grown, and the shoots of those plants were 66Zn-depleted relative to both their roots and bulk solution. Furthermore, Zn isotopes partition differently between different Zn-ligand complexes, so the form of Zn incorporated by organisms in the terrestrial biosphere plays a role in driving Zn isotope compositions of the organisms. In particular, based on ab initio calculations, Zn-phosphate complexes are expected to be isotopically heavier than Zn-citrates, Zn-malates and Zn-histidine complexes by 0.6 to 1‰.[89][90]
The discharge- and [Zn]-weighted average δ66Zn value of rivers throughout the world is +0.33‰.[73] In particular, the average δ66Zn values of the Kalix and Chang Jiang rivers are +0.64 and +0.56‰, respectively. The Amazon, Missouri and Brahmaputra rivers have average δ66Zn values near +0.30‰, and the average δ66Zn value of the Nile river is +0.21‰.[73]


In rocks and minerals
In general, δ66Zn values of various rocks and minerals do not appear to significantly vary. The δ66Zn value of bulk silicate Earth (BSE) is +0.28 + 0.05‰.[91] Fractionation of Zn isotopes by igneous processes is generally insignificant, and δ66Zn values of basalt fall within the range of +0.2 to +0.3‰,[68][69][70] encompassing the value for BSE. δ66Zn values of clay minerals from diverse environments and of diverse ages have been found to fall within the same range as basalts,[69] suggesting negligible fractionation between the basaltic precursors and sedimentary materials. Carbonates appear to be more 66Zn-enriched than other sedimentary and igneous rocks. For example, the δ66Zn value of a limestone core taken from the Central Pacific was +0.6‰ at the surface and increased to +1.2‰ with depth[92] The Zn isotopic compositions of various ores are not well-characterized, but smithsonites and sphalerites (Zn carbonates and Zn sulfides, respectively) collected from various localities in Europe had δ66Zn values ranging from -0.06 to +0.69‰, with smithsonite potentially slightly heavier by 0.3‰ than sphalerite.[68][69]
In the marine environment
Zn is an essential biological nutrient in the oceans, and its concentration is largely controlled by uptake by phytoplankton and remineralization. In addition to its critical role in many metalloenzymes (see Zinc Biology section), Zn is an important component of the carbonate shells of foraminifera[93] and siliceous frustules in diatoms.[94] The main inputs of Zn to the ocean are thought to be from rivers and dust.[73] In some photic zones in the ocean, Zn is a limiting nutrient for phytoplankton,[95] and thus its concentration in surface waters serves as one control on marine primary productivity. Zn concentrations are extremely low in the surface ocean (<0.1 nM) but are maximal at depth (~2 nM in the deep Atlantic; ~10 nM in the deep Pacific), indicating a deep regeneration cycle.[65][66] The deep/surface ratio of Zn is typically on the order of 100, significantly larger than that observed for Cu.
A multitude of complex processes fractionate Zn isotopes in the marine environment. As seen with copper isotopes, the bulk isotopic composition of zinc in the oceans (δ66Zn = +0.5‰) is heavier than that of the riverine input (δ66Zn = +0.3‰), reflecting both equilibrium, biological and other processes that affect Zn isotope ratios in the ocean.[73][66] In the surface ocean, phytoplankton preferentially uptake 64Zn, and as a result have average δ66Zn values of ~+0.16‰ (i.e., 0.34‰ lighter than the bulk ocean).[69] This preferential removal of 64Zn by photosynthetic marine organisms in the photic zone is most prominent in the spring and summer seasons when primary productivity is highest, and the seasonal variability of Zn isotope ratios is reflected in the δ66Zn values of settling materials, which are heavier (e.g., by ~+0.20‰ in the Atlantic Ocean) during spring and summer than during the colder seasons.[69] Additionally, the surface layers of FeMn-nodules are 66Zn enriched at high-latitudes (average δ66Zn = +1‰), while δ66Zn values of low-latitude samples are smaller and more variable (spanning +0.5 to +1‰).[69] This observation has been interpreted as due to high levels of Zn consumption and preferential uptake of 64Zn above the seasonal thermocline at high latitudes during warmer seasons, and transfer of this heavy δ66Zn signal to the settling sedimentary Fe-Mn hydroxides.[69]
Sources and sinks for Zn isotopes are further highlighted in the vertical profile of 66Zn/64Zn in the water column. In the upper 2,000 m of the Atlantic Ocean, δ66Zn values are highly variable near the surface (δ66Zn = +0.05 to +0.33‰)[66] due to biological uptake and other surface processes, then gradually increase to ~+0.50‰ at 2,000 m depth.[66] Potential sinks for light Zn isotopes, which enrich the residual bulk Zn isotope ratios in the ocean, include binding to and burial with sinking particulate matter, as well as Zn sulfide precipitation in buried sediments.[73][96] As a result of preferential burial of 64Zn over the heavier Zn isotopes, sediments in the ocean are generally isotopically lighter than that of bulk seawater. For example, δ66Zn values in 8 sedimentary cores from three different continental margins were depleted in 66Zn relative to the bulk ocean (δ66Zncores = -0.15 to +0.2‰), and furthermore the vertical profiles of δ66Zn values in the cores showed no downcore isotopic variability,[96] suggesting diagenesis does not significantly fractionate Zn isotopes.
Medicine
Zn isotopes may be useful as a tracer for breast cancer. Relative to non-cancerous patients, breast cancer patients are known to have significantly higher concentrations of Zn in their breast tissue, but lower concentrations in their blood serum and erythrocytes, due to overexpression of Zn transporters in breast cancer cells.[97][98][99][100] Consistent with these body-wide shifts in Zn homeostasis, δ66Zn values in breast cancer tumors of 5 patients were found to be anomalously light (varying from -0.9 to -0.6‰) relative to healthy tissue in 3 breast cancer patients and 1 healthy control (δ66Zn = -0.5 to -0.3‰).[101] In this study, δ66Zn values of blood and serum were not found to be significantly different between cancerous and non-cancerous patients, suggesting an unknown isotopically heavy pool of Zn must exist in cancer patients. Though results from this study are promising regarding the use of Zn isotope ratios as a biomarker for breast cancer, a mechanistic understanding of how Zn isotopes fractionate during tumor formation in breast cancer is still lacking. Fortunately, increasing attention is being devoted to the use of stable metal isotopes as tracers of cancer and other diseases, and the usefulness of these isotope systems in medical applications will become more apparent in the next few decades.
References
- Dauphas N, Rouxel O (2006). "Mass spectrometry and natural variations of iron isotopes". Mass Spectrometry Reviews. 25 (4): 515–50. Bibcode:2006MSRv...25..515D. doi:10.1002/mas.20078. PMID 16463281.
- Dauphas N, Janney PE, Mendybaev RA, Wadhwa M, Richter FM, Davis AM, et al. (October 2004). "Chromatographic separation and multicollection-ICPMS analysis of iron. Investigating mass-dependent and -independent isotope effects". Analytical Chemistry. 76 (19): 5855–63. doi:10.1021/ac0497095. PMID 15456307.
- Richter FM, Davis AM, DePaolo DJ, Watson EB (October 2003). "Isotope fractionation by chemical diffusion between molten basalt and rhyolite". Geochimica et Cosmochimica Acta. 67 (20): 3905–23. Bibcode:2003GeCoA..67.3905R. doi:10.1016/S0016-7037(03)00174-1.
- Anbar AD (2004-01-15). "Iron stable isotopes: beyond biosignatures". Earth and Planetary Science Letters. 217 (3–4): 223–236. Bibcode:2004E&PSL.217..223A. doi:10.1016/S0012-821X(03)00572-7.
- Johnson CM, Beard BL, Beukes NJ, Klein C, O'Leary JM (February 2003). "Ancient geochemical cycling in the Earth as inferred from Fe isotope studies of banded iron formations from the Transvaal Craton". Contributions to Mineralogy and Petrology. 144 (5): 523–47. doi:10.1007/s00410-002-0418-x. S2CID 130262522.
- Bruland KW, Lohan MC (2003). "Controls of Trace Metals in Seawater.". In Elderfield H (ed.). The Oceans and Marine Geochemistry. Oxford: Elsevier-Pergamon. pp. 23–47. ISBN 978-0-08-045101-5.
- Jaouen, Klervia; Pons, Marie-Laure; Balter, Vincent (2013-07-15). "Iron, copper and zinc isotopic fractionation up mammal trophic chains". Earth and Planetary Science Letters. 374: 164–172. Bibcode:2013E&PSL.374..164J. doi:10.1016/j.epsl.2013.05.037. ISSN 0012-821X.
- Poitrasson F, Halliday AN, Lee DC, Levasseur S, Teutsch N (July 2004). "Iron isotope differences between Earth, Moon, Mars and Vesta as possible records of contrasted accretion mechanisms". Earth and Planetary Science Letters. 223 (3–4): 253–66. Bibcode:2004E&PSL.223..253P. doi:10.1016/j.epsl.2004.04.032.
- Dauphas N, John SG, Rouxel O (January 2017). "Iron Isotope Systematics". Reviews in Mineralogy and Geochemistry. 82 (1): 415–510. Bibcode:2017RvMG...82..415D. doi:10.2138/rmg.2017.82.11.
- Bullen TD, White AF, Childs CW, Vivit DV, Schulz MS (August 2001). "Demonstration of significant abiotic iron isotope fractionation in nature". Geology. 29 (8): 699–702. Bibcode:2001Geo....29..699B. doi:10.1130/0091-7613(2001)029<0699:DOSAII>2.0.CO;2.
- Dauphas N (2007). "Diffusion-driven kinetic isotope effect of Fe and Ni during formation of the Widmanstätten pattern". Meteoritics & Planetary Science. 42 (9): 1597–1613. Bibcode:2007M&PS...42.1597D. doi:10.1111/j.1945-5100.2007.tb00593.x.
- Richter FM, Watson EB, Mendybaev R, Dauphas N, Georg B, Watkins J, Valley J (July 2009). "Isotopic fractionation of the major elements of molten basalt by chemical and thermal diffusion". Geochimica et Cosmochimica Acta. 73 (14): 4250–63. Bibcode:2009GeCoA..73.4250R. doi:10.1016/j.gca.2009.04.011.
- Rodushkin I, Stenberg A, Andrén H, Malinovsky D, Baxter DC (April 2004). "Isotopic fractionation during diffusion of transition metal ions in solution". Analytical Chemistry. 76 (7): 2148–51. doi:10.1021/ac035296g. PMID 15053683.
- Roskosz M, Luais B, Watson H, Toplis MJ, Alexander CM, Mysen BO (December 2005). "High-Temperature Fractionation of Iron Isotopes During Metal Segregation From a Silicate Melt: Experimental Study of Kinetic and Equilibrium Fractionation". InAGU Fall Meeting.
- Beard BL, Johnson CM, Cox L, Sun H, Nealson KH, Aguilar C (September 1999). "Iron isotope biosignatures". Science. 285 (5435): 1889–92. doi:10.1126/science.285.5435.1889. PMID 10489362.
- Icopini GA, Anbar AD, Ruebush SS, Tien M, Brantley SL (March 2004). "Iron isotope fractionation during microbial reduction of iron: the importance of adsorption". Geology. 32 (3): 205–8. Bibcode:2004Geo....32..205I. doi:10.1130/G20184.1.
- Beard BL, Johnson CM, Skulan JL, Nealson KH, Cox L, Sun H (April 2003). "Application of Fe isotopes to tracing the geochemical and biological cycling of Fe". Chemical Geology. 195 (1–4): 87–117. Bibcode:2003ChGeo.195...87B. doi:10.1016/S0009-2541(02)00390-X.
- Crosby HA, Johnson CM, Roden EE, Beard BL (September 2005). "Coupled Fe(II)-Fe(III) electron and atom exchange as a mechanism for Fe isotope fractionation during dissimilatory iron oxide reduction". Environmental Science & Technology. 39 (17): 6698–704. Bibcode:2005EnST...39.6698C. doi:10.1021/es0505346. PMID 16190229.
- Johnson CM, Roden EE, Welch SA, Beard BL (February 2005). "Experimental constraints on Fe isotope fractionation during magnetite and Fe carbonate formation coupled to dissimilatory hydrous ferric oxide reduction". Geochimica et Cosmochimica Acta. 69 (4): 963–93. Bibcode:2005GeCoA..69..963J. doi:10.1016/j.gca.2004.06.043.
- Balci N, Bullen TD, Witte-Lien K, Shanks WC, Motelica M, Mandernack KW (February 2006). "Iron isotope fractionation during microbially stimulated Fe (II) oxidation and Fe (III) precipitation". Geochimica et Cosmochimica Acta. 70 (3): 622–39. Bibcode:2006GeCoA..70..622B. doi:10.1016/j.gca.2005.09.025.
- Croal LR, Johnson CM, Beard BL, Newman DK (2004). "Iron isotope fractionation by anoxygenic Fe (II)-phototrophic bacteria". Geochim Cosmochim Acta. 68: 1227–42. doi:10.1016/j.gca.2003.09.011.
- Mandernack KW, Bazylinski DA, Shanks WC, Bullen TD (September 1999). "Oxygen and iron isotope studies of magnetite produced by magnetotactic bacteria". Science. 285 (5435): 1892–6. doi:10.1126/science.285.5435.1892. PMID 10489363.
- Schoffman H, Lis H, Shaked Y, Keren N (2016-08-18). "Iron-Nutrient Interactions within Phytoplankton". Frontiers in Plant Science. 7: 1223. doi:10.3389/fpls.2016.01223. PMC 4989028. PMID 27588022.
- Radic A, Lacan F, Murray JW (June 2011). "Iron isotopes in the seawater of the equatorial Pacific Ocean: New constraints for the oceanic iron cycle" (PDF). Earth and Planetary Science Letters. 306 (1–2): 1–10. Bibcode:2011E&PSL.306....1R. doi:10.1016/j.epsl.2011.03.015.
- Walczyk T, von Blanckenburg F (April 2005). "Deciphering the iron isotope message of the human body". International Journal of Mass Spectrometry. 242 (2–3): 117–34. Bibcode:2005IJMSp.242..117W. doi:10.1016/j.ijms.2004.12.028.
- Walczyk T, von Blanckenburg F (March 2002). "Natural iron isotope variations in human blood". Science. 295 (5562): 2065–6. Bibcode:2002Sci...295.2065W. doi:10.1126/science.1069389. PMID 11896276. S2CID 45665175.
- Dauphas N, van Zuilen M, Wadhwa M, Davis AM, Marty B, Janney PE (December 2004). "Clues from Fe isotope variations on the origin of early Archean BIFs from Greenland". Science. 306 (5704): 2077–80. Bibcode:2004Sci...306.2077D. doi:10.1126/science.1104639. PMID 15604404. S2CID 35078356.
- Beard BL, Johnson CM (June 1999). "High precision iron isotope measurements of terrestrial and lunar materials". Geochimica et Cosmochimica Acta. 63 (11–12): 1653–60. Bibcode:1999GeCoA..63.1653B. doi:10.1016/S0016-7037(99)00089-7.
- Lacan, F.; Radic, A.; Jeandel, C.; Poitrasson, F.; Sarthou, G.; Pradoux, C.; Freydier, R. (31 December 2008). "Measurement of the isotopic composition of dissolved iron in the open ocean". Geophysical Research Letters. 35 (24): L24610. Bibcode:2008GeoRL..3524610L. doi:10.1029/2008GL035841. S2CID 18442017.
- John SG, Adkins J (June 2012). "The vertical distribution of iron stable isotopes in the North Atlantic near Bermuda" (PDF). Global Biogeochemical Cycles. 26 (2): n/a. Bibcode:2012GBioC..26.2034J. doi:10.1029/2011GB004043. S2CID 29125362.
- Conway TM, John SG (July 2014). "Quantification of dissolved iron sources to the North Atlantic Ocean". Nature. 511 (7508): 212–5. Bibcode:2014Natur.511..212C. doi:10.1038/nature13482. PMID 25008528. S2CID 4462901.
- Labatut M, Lacan F, Pradoux C, Chmeleff J, Radic A, Murray JW, Poitrasson F, Johansen AM, Thil F (October 2014). "Iron sources and dissolved‐particulate interactions in the seawater of the Western Equatorial Pacific, iron isotope perspectives" (PDF). Global Biogeochemical Cycles. 28 (10): 1044–1065. Bibcode:2014GBioC..28.1044L. doi:10.1002/2014GB004928. ISSN 1944-9224. S2CID 34615343.
- Radic A, Lacan F, Murray JW (2011-06-01). "Iron isotopes in the seawater of the equatorial Pacific Ocean: New constraints for the oceanic iron cycle" (PDF). Earth and Planetary Science Letters. 306 (1–2): 1–10. Bibcode:2011E&PSL.306....1R. doi:10.1016/j.epsl.2011.03.015. ISSN 0012-821X.
- Wiesli RA, Beard BL, Taylor LA, Johnson CM (December 2003). "Space weathering processes on airless bodies: Fe isotope fractionation in the lunar regolith 1". Earth and Planetary Science Letters. 216 (4): 457–465. Bibcode:2003E&PSL.216..457W. doi:10.1016/S0012-821X(03)00552-1.
- Johnson CM, Beard BL, Klein C, Beukes NJ, Roden EE (January 2008). "Iron isotopes constrain biologic and abiologic processes in banded iron formation genesis". Geochimica et Cosmochimica Acta. 72 (1): 151–69. Bibcode:2008GeCoA..72..151J. doi:10.1016/j.gca.2007.10.013.
- Kappler A, Pasquero C, Konhauser KO, Newman DK (November 2005). "Deposition of banded iron formations by anoxygenic phototrophic Fe (II)-oxidizing bacteria". Geology. 33 (11): 865–8. Bibcode:2005Geo....33..865K. doi:10.1130/G21658.1.
- Boyd PW, Ellwood MJ (2010-09-26). "The biogeochemical cycle of iron in the ocean". Nature Geoscience. 3 (10): 675–682. Bibcode:2010NatGe...3..675B. doi:10.1038/ngeo964. hdl:1885/55479.
- Abadie C, Lacan F, Radic A, Pradoux C, Poitrasson F (January 2017). "Iron isotopes reveal distinct dissolved iron sources and pathways in the intermediate versus deep Southern Ocean". Proceedings of the National Academy of Sciences of the United States of America. 114 (5): 858–863. Bibcode:2017PNAS..114..858A. doi:10.1073/pnas.1603107114. PMC 5293069. PMID 28096366.
- Revels BN, Ohnemus DC, Lam PJ, Conway TM, John SG (June 2015). "The isotopic signature and distribution of particulate iron in the North Atlantic Ocean". Deep-Sea Research Part II: Topical Studies in Oceanography. 116: 321–31. Bibcode:2015DSRII.116..321R. doi:10.1016/j.dsr2.2014.12.004.
- Fitzsimmons JN, Conway TM, Lee JM, Kayser R, Thyng KM, John SG, Boyle EA (October 2016). "Dissolved iron and iron isotopes in the southeastern Pacific Ocean". Global Biogeochemical Cycles. 30 (10): 1372–95. Bibcode:2016GBioC..30.1372F. doi:10.1002/2015GB005357.
- O'Neil JR (1986-12-31). "Chapter 1. Theoretical and Experimental Aspects of Isotopic Fractionation". In Valley JW, Taylor HP, O'Neil JR (eds.). Stable Isotopes in High Temperature Geological Processes. De Gruyter. pp. 1–40. doi:10.1515/9781501508936-006. ISBN 978-1-5015-0893-6.
- Nealson KH, Cox BL (June 2002). "Microbial metal-ion reduction and Mars: extraterrestrial expectations?". Current Opinion in Microbiology. 5 (3): 296–300. doi:10.1016/S1369-5274(02)00326-0. PMID 12057684.
- Thomas-Keprta KL, Clemett SJ, Bazylinski DA, Kirschvink JL, McKay DS, Wentworth SJ, et al. (August 2002). "Magnetofossils from ancient Mars: a robust biosignature in the martian meteorite ALH84001". Applied and Environmental Microbiology. 68 (8): 3663–72. Bibcode:2002ApEnM..68.3663T. doi:10.1128/AEM.68.8.3663-3672.2002. PMC 123990. PMID 12147458.
- Winter WE, Bazydlo LA, Harris NS (Spring 2014). "The molecular biology of human iron metabolism". Laboratory Medicine. 45 (2): 92–102. doi:10.1309/lmf28s2gimxnwhmm. PMID 24868988.
- Bullen TD, Walczyk T (2009-12-01). "Environmental and Biomedical Applications of Natural Metal Stable Isotope Variations". Elements. 5 (6): 381–385. doi:10.2113/gselements.5.6.381. ISSN 1811-5209.
- Zhu XK, Guo Y, Williams RJ, O'Nions RK, Matthews A, Belshaw NS, et al. (June 2002). "Mass fractionation processes of transition metal isotopes". Earth and Planetary Science Letters. 200 (1–2): 47–62. Bibcode:2002E&PSL.200...47Z. doi:10.1016/S0012-821X(02)00615-5. hdl:1887/3608167.
- Rouxel OD (2002). Géochimie isotopique des métaux (Fe, Cu, Pb) et des metalloïdes (S, Se) dans la croûte océanique [Isotopic geochemistry of metals (Fe, Cu, Pb) and metalloids (S, Se) in the oceanic crust.] (Ph.D. thesis) (in French). Nancy: Institut National Polytechnique de Lorraine.
- Fujii T, Moynier F, Abe M, Nemoto K, Albarède F (June 2013). "Copper isotope fractionation between aqueous compounds relevant to low temperature geochemistry and biology". Geochimica et Cosmochimica Acta. 110: 29–44. Bibcode:2013GeCoA.110...29F. doi:10.1016/j.gca.2013.02.007. hdl:2433/173360.
- McDonough WF, Sun SS (March 1995). "The composition of the Earth". Chemical Geology. 120 (3–4): 223–53. Bibcode:1995ChGeo.120..223M. doi:10.1016/0009-2541(94)00140-4.
- Albarède F (January 2004). "The stable isotope geochemistry of copper and zinc". Reviews in Mineralogy and Geochemistry. 55 (1): 409–27. Bibcode:2004RvMG...55..409A. doi:10.2138/gsrmg.55.1.409.
- Maréchal CN, Télouk P, Albarède F (April 1999). "Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry". Chemical Geology. 156 (1–4): 251–73. Bibcode:1999ChGeo.156..251M. doi:10.1016/S0009-2541(98)00191-0.
- Shields WR, Murphy TJ, Garner EL (1964). "Absolute Isotopic Abundance Ratio and the Atomic Weight of a Reference Sample of Copper". Journal of Research of the National Bureau of Standards Section A. 68A (6): 589–592. doi:10.6028/jres.068A.056. PMC 6592378. PMID 31834740.
- Shields WR, Goldich SS, Garner EL, Murphy TJ (January 1965). "Natural variations in the abundance ratio and the atomic weight of copper". Journal of Geophysical Research. 70 (2): 479–91. Bibcode:1965JGR....70..479S. doi:10.1029/JZ070i002p00479.
- Albarède F (August 2015). "Metal stable isotopes in the human body: a tribute of geochemistry to medicine". Elements. 11 (4): 265–9. doi:10.2113/gselements.11.4.265.
- Albarède F, Telouk P, Lamboux A, Jaouen K, Balter V (September 2011). "Isotopic evidence of unaccounted for Fe and Cu erythropoietic pathways". Metallomics. 3 (9): 926–33. Bibcode:2011AGUFM.B54D..06A. doi:10.1039/c1mt00025j. PMID 21789323.
- Balter V, Nogueira da Costa A, Bondanese VP, Jaouen K, Lamboux A, Sangrajrang S, et al. (January 2015). "Natural variations of copper and sulfur stable isotopes in blood of hepatocellular carcinoma patients". Proceedings of the National Academy of Sciences of the United States of America. 112 (4): 982–5. Bibcode:2015PNAS..112..982B. doi:10.1073/pnas.1415151112. PMC 4313854. PMID 25583489.
- Jaouen K, Balter V (February 2014). "Menopause effect on blood Fe and Cu isotope compositions". American Journal of Physical Anthropology. 153 (2): 280–5. doi:10.1002/ajpa.22430. PMID 24263674.
- Bigalke M, Weyer S, Wilcke W (January 2010). "Stable copper isotopes: A novel tool to trace copper behavior in hydromorphic soils". Soil Science Society of America Journal. 74 (1): 60–73. Bibcode:2010SSASJ..74...60B. doi:10.2136/sssaj2008.0377.
- Kříbek B, Šípková A, Ettler V, Mihaljevič M, Majer V, Knésl I, et al. (August 2018). "Variability of the copper isotopic composition in soil and grass affected by mining and smelting in Tsumeb, Namibia". Chemical Geology. 493: 121–35. Bibcode:2018ChGeo.493..121K. doi:10.1016/j.chemgeo.2018.05.035. S2CID 134818140.
- Weiss DJ, Mason TF, Zhao FJ, Kirk GJ, Coles BJ, Horstwood MS (March 2005). "Isotopic discrimination of zinc in higher plants" (PDF). The New Phytologist. 165 (3): 703–10. doi:10.1111/j.1469-8137.2004.01307.x. PMID 15720681. S2CID 30007608.
- Jouvin D, Weiss DJ, Mason TF, Bravin MN, Louvat P, Zhao F, et al. (March 2012). "Stable isotopes of Cu and Zn in higher plants: evidence for Cu reduction at the root surface and two conceptual models for isotopic fractionation processes". Environmental Science & Technology. 46 (5): 2652–60. Bibcode:2012EnST...46.2652J. doi:10.1021/es202587m. PMID 22296233.
- Weinstein C, Moynier F, Wang K, Paniello R, Foriel J, Catalano J, Pichat S (July 2011). "Isotopic fractionation of Cu in plants". Chemical Geology. 286 (3–4): 266–71. Bibcode:2011ChGeo.286..266W. doi:10.1016/j.chemgeo.2011.05.010.
- Vance D, Archer C, Bermin J, Perkins J, Statham PJ, Lohan MC, Ellwood MJ, Mills RA (September 2008). "The copper isotope geochemistry of rivers and the oceans". Earth and Planetary Science Letters. 274 (1–2): 204–13. Bibcode:2008E&PSL.274..204V. doi:10.1016/j.epsl.2008.07.026.
- Bruland KW (April 1980). "Oceanographic distributions of cadmium, zinc, nickel, and copper in the North Pacific". Earth and Planetary Science Letters. 47 (2): 176–98. Bibcode:1980E&PSL..47..176B. doi:10.1016/0012-821X(80)90035-7.
- Boyle EA, John S, Abouchami W, Adkins JF, Echegoyen-Sanz Y, Ellwood M, et al. (September 2012). "GEOTRACES IC1 (BATS) contamination‐prone trace element isotopes Cd, Fe, Pb, Zn, Cu, and Mo intercalibration". Limnology and Oceanography: Methods. 10 (9): 653–65. doi:10.4319/lom.2012.10.653. hdl:1912/6632.
- Liu SA, Huang J, Liu J, Wörner G, Yang W, Tang YJ, et al. (October 2015). "Copper isotopic composition of the silicate Earth. Earth and Planetary Science Letters". 427: 95–103. doi:10.1016/j.epsl.2015.06.061.
{{cite journal}}: Cite journal requires|journal=(help) - Maréchal C (1998). Géochimie des isotopes du Cuivre et du Zinc. Méthode, variabilités naturelles, et application océanographique [Geochemistry of Copper and Zinc isotopes. Method, natural variability, and oceanographic application] (Ph.D. thesis) (in French). Saint-Martin-d'Hères: Université de Grenoble 1. p. 253.
- Maréchal CN, Nicolas E, Douchet C, Albarède F (May 2000). "Abundance of zinc isotopes as a marine biogeochemical tracer". Geochemistry, Geophysics, Geosystems. 1 (5): 999GC–000029. Bibcode:2000GGG.....1.1015M. doi:10.1029/1999GC000029.
- Ben OD, Luck JM, Tchalikian JM, Albarède F (2003). "Cu–Zn isotope systematics in terrestrial basalts". Geophysical. Res. Abstracts. 5: 9669. Bibcode:2003EAEJA.....9669B.
- Rouxel O, Fouquet Y, Ludden JN (May 2004). "Copper isotope systematics of the Lucky Strike, Rainbow, and Logatchev sea-floor hydrothermal fields on the Mid-Atlantic Ridge". Economic Geology. 99 (3): 585–600. Bibcode:2004EcGeo..99..585R. doi:10.2113/gsecongeo.99.3.585.
- Larson PB, Maher K, Ramos FC, Chang Z, Gaspar M, Meinert LD (November 2003). "Copper isotope ratios in magmatic and hydrothermal ore-forming environments". Chemical Geology. 201 (3–4): 337–50. Bibcode:2003ChGeo.201..337L. doi:10.1016/j.chemgeo.2003.08.006.
- Little SH, Vance D, Walker-Brown C, Landing WM (January 2014). "The oceanic mass balance of copper and zinc isotopes, investigated by analysis of their inputs, and outputs to ferromanganese oxide sediments". Geochimica et Cosmochimica Acta. 125: 673–93. Bibcode:2014GeCoA.125..673L. doi:10.1016/j.gca.2013.07.046.
- Goode CA (1991). "Copper and disease". In Linder MC (ed.). Biochemistry of Copper. New York: Springer. pp. 331–366. ISBN 978-0-306-43658-1.
- Albarede F, Télouk P, Balter V, Bondanese VP, Albalat E, Oger P, Bonaventura P, Miossec P, Fujii T (October 2016). "Medical applications of Cu, Zn, and S isotope effects". Metallomics: Integrated Biometal Science. 8 (10): 1056–1070. doi:10.1039/c5mt00316d. PMID 27513195.
- Andreini C, Banci L, Bertini I, Rosato A (January 2006). "Counting the zinc-proteins encoded in the human genome". Journal of Proteome Research. 5 (1): 196–201. doi:10.1021/pr050361j. PMID 16396512.
- Maret W (February 2010). "Metalloproteomics, metalloproteomes, and the annotation of metalloproteins". Metallomics: Integrated Biometal Science. 2 (2): 117–25. doi:10.1039/b915804a. PMID 21069142.
- Krężel A, Maret W (December 2016). "The biological inorganic chemistry of zinc ions". Archives of Biochemistry and Biophysics. 611: 3–19. doi:10.1016/j.abb.2016.04.010. PMC 5120989. PMID 27117234.
- Krężel A, Maret W (November 2006). "Zinc-buffering capacity of a eukaryotic cell at physiological pZn". Journal of Biological Inorganic Chemistry. 11 (8): 1049–62. doi:10.1007/s00775-006-0150-5. PMID 16924557. S2CID 1200984.
- Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M (October 2009). "Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis". Nature Methods. 6 (10): 737–40. doi:10.1038/nmeth.1368. PMC 6101214. PMID 19718032.
- John SG, Geis RW, Saito MA, Boyle EA (November 2007). "Zinc isotope fractionation during high‐affinity and low‐affinity zinc transport by the marine diatom Thalassiosira oceanica". Limnology and Oceanography. 52 (6): 2710–4. Bibcode:2007LimOc..52.2710J. doi:10.4319/lo.2007.52.6.2710.
- Gélabert A, Pokrovsky OS, Viers J, Schott J, Boudou A, Feurtet-Mazel A (February 2006). "Interaction between zinc and freshwater and marine diatom species: surface complexation and Zn isotope fractionation". Geochimica et Cosmochimica Acta. 70 (4): 839–57. Bibcode:2006GeCoA..70..839G. doi:10.1016/j.gca.2005.10.026.
- Kafantaris FC, Borrok DM (February 2014). "Zinc isotope fractionation during surface adsorption and intracellular incorporation by bacteria". Chemical Geology. 366: 42–51. Bibcode:2014ChGeo.366...42K. doi:10.1016/j.chemgeo.2013.12.007.
- Stanley Jr JK, Byrne RH (March 1990). "Inorganic complexation of Zinc (II) in seawater". Geochimica et Cosmochimica Acta. 54 (3): 753–60. Bibcode:1990GeCoA..54..753S. doi:10.1016/0016-7037(90)90370-Z.
- Millero FJ (1996). Chemical Oceanography. Marine Science Series (2nd ed.). Boca Raton: CRC Press. ISBN 978-0-8493-8423-3.
- Costas-Rodríguez M, Anoshkina Y, Lauwens S, Van Vlierberghe H, Delanghe J, Vanhaecke F (March 2015). "Isotopic analysis of Cu in blood serum by multi-collector ICP-mass spectrometry: a new approach for the diagnosis and prognosis of liver cirrhosis?". Metallomics: Integrated Biometal Science. 7 (3): 491–8. doi:10.1039/c4mt00319e. PMID 25644127.
- Van Heghe L, Engström E, Rodushkin I, Cloquet C, Vanhaecke F (2012). "Isotopic analysis of the metabolically relevant transition metals Cu, Fe and Zn in human blood from vegetarians and omnivores using multi-collector ICP-mass spectrometry". Journal of Analytical Atomic Spectrometry. 27 (8): 1327–34. doi:10.1039/C2JA30070B.
- Opfergelt S, Cornélis JT, Houben D, Givron C, Burton KW, Mattielli N (September 2017). "The influence of weathering and soil organic matter on Zn isotopes in soils". Chemical Geology. 466: 140–8. Bibcode:2017ChGeo.466..140O. doi:10.1016/j.chemgeo.2017.06.002. hdl:2268/215015.
- Fujii T, Albarède F (2012). "Ab initio calculation of the Zn isotope effect in phosphates, citrates, and malates and applications to plants and soil". PLOS ONE. 7 (2): e30726. Bibcode:2012PLoSO...730726F. doi:10.1371/journal.pone.0030726. PMC 3281869. PMID 22363478.
- Fujii T, Moynier F, Blichert-Toft J, Albarède F (September 2014). "Density functional theory estimation of isotope fractionation of Fe, Ni, Cu, and Zn among species relevant to geochemical and biological environments". Geochimica et Cosmochimica Acta. 140: 553–76. Bibcode:2014GeCoA.140..553F. doi:10.1016/j.gca.2014.05.051. hdl:2433/189321.
- Chen H, Savage PS, Teng FZ, Helz RT, Moynier F (May 2013). "Zinc isotope fractionation during magmatic differentiation and the isotopic composition of the bulk Earth". Earth and Planetary Science Letters. 369: 34–42. Bibcode:2013E&PSL.369...34C. doi:10.1016/j.epsl.2013.02.037.
- Pichat S, Douchet C, Albarède F (May 2003). "Zinc isotope variations in deep-sea carbonates from the eastern equatorial Pacific over the last 175 ka". Earth and Planetary Science Letters. 210 (1–2): 167–78. Bibcode:2003E&PSL.210..167P. doi:10.1016/S0012-821X(03)00106-7.
- Boyle EA (March 1981). "Cadmium, zinc, copper, and barium in foraminifera tests". Earth and Planetary Science Letters. 53 (1): 11–35. Bibcode:1981E&PSL..53...11B. doi:10.1016/0012-821X(81)90022-4.
- Collier R, Edmond J (January 1984). "The trace element geochemistry of marine biogenic particulate matter". Progress in Oceanography. 13 (2): 113–99. Bibcode:1984PrOce..13..113C. doi:10.1016/0079-6611(84)90008-9. hdl:1721.1/51420.
- Price NM, Morel FM (April 1990). "Cadmium and cobalt substitution for zinc in a marine diatom". Nature. 344 (6267): 658–60. Bibcode:1990Natur.344..658P. doi:10.1038/344658a0. S2CID 4242770.
- Little SH, Vance D, McManus J, Severmann S (March 2016). "Key role of continental margin sediments in the oceanic mass balance of Zn and Zn isotopes". Geology. 44 (3): 207–10. Bibcode:2016Geo....44..207L. doi:10.1130/G37493.1. hdl:20.500.11850/113977.
- Margalioth EJ, Schenker JG, Chevion M (September 1983). "Copper and zinc levels in normal and malignant tissues". Cancer. 52 (5): 868–72. doi:10.1002/1097-0142(19830901)52:5<868::aid-cncr2820520521>3.0.co;2-k. PMID 6871828.
- Alam S, Kelleher SL (August 2012). "Cellular mechanisms of zinc dysregulation: a perspective on zinc homeostasis as an etiological factor in the development and progression of breast cancer". Nutrients. 4 (8): 875–903. doi:10.3390/nu4080875. PMC 3448077. PMID 23016122.
- Lopez V, Foolad F, Kelleher SL (May 2011). "ZnT2-overexpression represses the cytotoxic effects of zinc hyper-accumulation in malignant metallothionein-null T47D breast tumor cells". Cancer Letters. 304 (1): 41–51. doi:10.1016/j.canlet.2011.01.027. PMID 21353385.
- Tinoco-Veras CM, Bezerra Sousa MS, da Silva BB, Franciscato Cozzolino SM, Viana Pires L, Coelho Pimentel JA, do Nascimento-Nogueira N, do Nascimento-Marreiro D (2011). "Analysis of plasma and erythrocyte zinc levels in premenopausal women with breast cancer". Nutricion Hospitalaria. 26 (2): 293–7. doi:10.1590/S0212-16112011000200008 (inactive 1 August 2023). PMID 21666965.
{{cite journal}}: CS1 maint: DOI inactive as of August 2023 (link) - Larner F, Woodley LN, Shousha S, Moyes A, Humphreys-Williams E, Strekopytov S, Halliday AN, Rehkämper M, Coombes RC (January 2015). "Zinc isotopic compositions of breast cancer tissue". Metallomics: Integrated Biometal Science. 7 (1): 112–7. doi:10.1039/c4mt00260a. PMID 25489714.