Epistasis
Epistasis is a phenomenon in genetics in which the effect of a gene mutation is dependent on the presence or absence of mutations in one or more other genes, respectively termed modifier genes. In other words, the effect of the mutation is dependent on the genetic background in which it appears.[2] Epistatic mutations therefore have different effects on their own than when they occur together. Originally, the term epistasis specifically meant that the effect of a gene variant is masked by that of a different gene.[3]

The concept of epistasis originated in genetics in 1907[4] but is now used in biochemistry, computational biology and evolutionary biology. The phenomenon arises due to interactions, either between genes (such as mutations also being needed in regulators of gene expression) or within them (multiple mutations being needed before the gene loses function), leading to non-linear effects. Epistasis has a great influence on the shape of evolutionary landscapes, which leads to profound consequences for evolution and for the evolvability of phenotypic traits.
History
Understanding of epistasis has changed considerably through the history of genetics and so too has the use of the term. The term was first used by William Bateson and his collaborators Florence Durham and Muriel Wheldale Onslow.[4] In early models of natural selection devised in the early 20th century, each gene was considered to make its own characteristic contribution to fitness, against an average background of other genes. Some introductory courses still teach population genetics this way. Because of the way that the science of population genetics was developed, evolutionary geneticists have tended to think of epistasis as the exception. However, in general, the expression of any one allele depends in a complicated way on many other alleles.
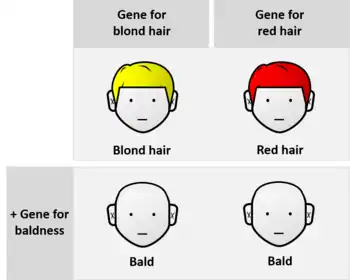
In classical genetics, if genes A and B are mutated, and each mutation by itself produces a unique phenotype but the two mutations together show the same phenotype as the gene A mutation, then gene A is epistatic and gene B is hypostatic. For example, the gene for total baldness is epistatic to the gene for brown hair. In this sense, epistasis can be contrasted with genetic dominance, which is an interaction between alleles at the same gene locus. As the study of genetics developed, and with the advent of molecular biology, epistasis started to be studied in relation to quantitative trait loci (QTL) and polygenic inheritance.
The effects of genes are now commonly quantifiable by assaying the magnitude of a phenotype (e.g. height, pigmentation or growth rate) or by biochemically assaying protein activity (e.g. binding or catalysis). Increasingly sophisticated computational and evolutionary biology models aim to describe the effects of epistasis on a genome-wide scale and the consequences of this for evolution.[5][6][7] Since identification of epistatic pairs is challenging both computationally and statistically, some studies try to prioritize epistatic pairs.[8][9]
Classification
Terminology about epistasis can vary between scientific fields. Geneticists often refer to wild type and mutant alleles where the mutation is implicitly deleterious and may talk in terms of genetic enhancement, synthetic lethality and genetic suppressors. Conversely, a biochemist may more frequently focus on beneficial mutations and so explicitly state the effect of a mutation and use terms such as reciprocal sign epistasis and compensatory mutation.[16] Additionally, there are differences when looking at epistasis within a single gene (biochemistry) and epistasis within a haploid or diploid genome (genetics). In general, epistasis is used to denote the departure from 'independence' of the effects of different genetic loci. Confusion often arises due to the varied interpretation of 'independence' among different branches of biology.[17] The classifications below attempt to cover the various terms and how they relate to one another.
Additivity
Two mutations are considered to be purely additive if the effect of the double mutation is the sum of the effects of the single mutations. This occurs when genes do not interact with each other, for example by acting through different metabolic pathways. Simply, additive traits were studied early on in the history of genetics, however they are relatively rare, with most genes exhibiting at least some level of epistatic interaction.[18][19]
Magnitude epistasis
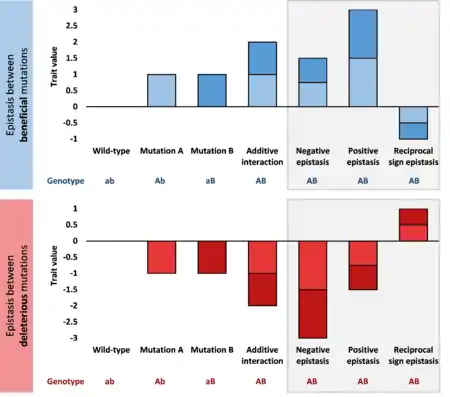
When the double mutation has a fitter phenotype than expected from the effects of the two single mutations, it is referred to as positive epistasis. Positive epistasis between beneficial mutations generates greater improvements in function than expected.[10][11] Positive epistasis between deleterious mutations protects against the negative effects to cause a less severe fitness drop.[13]
Conversely, when two mutations together lead to a less fit phenotype than expected from their effects when alone, it is called negative epistasis.[20][21] Negative epistasis between beneficial mutations causes smaller than expected fitness improvements, whereas negative epistasis between deleterious mutations causes greater-than-additive fitness drops.[12]
Independently, when the effect on fitness of two mutations is more radical than expected from their effects when alone, it is referred to as synergistic epistasis. The opposite situation, when the fitness difference of the double mutant from the wild type is smaller than expected from the effects of the two single mutations, it is called antagonistic epistasis.[15] Therefore, for deleterious mutations, negative epistasis is also synergistic, while positive epistasis is antagonistic; conversely, for advantageous mutations, positive epistasis is synergistic, while negative epistasis is antagonistic.
The term genetic enhancement is sometimes used when a double (deleterious) mutant has a more severe phenotype than the additive effects of the single mutants. Strong positive epistasis is sometimes referred to by creationists as irreducible complexity (although most examples are misidentified).
Sign epistasis
Sign epistasis[22] occurs when one mutation has the opposite effect when in the presence of another mutation. This occurs when a mutation that is deleterious on its own can enhance the effect of a particular beneficial mutation.[17] For example, a large and complex brain is a waste of energy without a range of sense organs, but sense organs are made more useful by a large and complex brain that can better process the information. If a fitness landscape has no sign epistasis then it is called smooth.
At its most extreme, reciprocal sign epistasis[23] occurs when two deleterious genes are beneficial when together. For example, producing a toxin alone can kill a bacterium, and producing a toxin exporter alone can waste energy, but producing both can improve fitness by killing competing organisms. If a fitness landscape has sign epistasis but no reciprocal sign epistasis then it is called semismooth.[24]
Reciprocal sign epistasis also leads to genetic suppression whereby two deleterious mutations are less harmful together than either one on its own, i.e. one compensates for the other. A clear example of genetic suppression was the demonstration that in the assembly of bacteriophage T4 two deleterious mutations, each causing a deficiency in the level of a different morphogenetic protein, could interact positively.[25] If a mutation causes a reduction in a particular structural component, this can bring about an imbalance in morphogenesis and loss of viable virus progeny, but production of viable progeny can be restored by a second (suppressor) mutation in another morphogenetic component that restores the balance of protein components.
The term genetic suppression can also apply to sign epistasis where the double mutant has a phenotype intermediate between those of the single mutants, in which case the more severe single mutant phenotype is suppressed by the other mutation or genetic condition. For example, in a diploid organism, a hypomorphic (or partial loss-of-function) mutant phenotype can be suppressed by knocking out one copy of a gene that acts oppositely in the same pathway. In this case, the second gene is described as a "dominant suppressor" of the hypomorphic mutant; "dominant" because the effect is seen when one wild-type copy of the suppressor gene is present (i.e. even in a heterozygote). For most genes, the phenotype of the heterozygous suppressor mutation by itself would be wild type (because most genes are not haplo-insufficient), so that the double mutant (suppressed) phenotype is intermediate between those of the single mutants.
In non reciprocal sign epistasis, fitness of the mutant lies in the middle of that of the extreme effects seen in reciprocal sign epistasis.
When two mutations are viable alone but lethal in combination, it is called Synthetic lethality or unlinked non-complementation.[26]
Haploid organisms
In a haploid organism with genotypes (at two loci) ab, Ab, aB or AB, we can think of different forms of epistasis as affecting the magnitude of a phenotype upon mutation individually (Ab and aB) or in combination (AB).
| Interaction type | ab | Ab | aB | AB | |
| No epistasis (additive) | 0 | 1 | 1 | 2 | AB = Ab + aB + ab |
| Positive (synergistic) epistasis | 0 | 1 | 1 | 3 | AB > Ab + aB + ab |
| Negative (antagonistic) epistasis | 0 | 1 | 1 | 1 | AB < Ab + aB + ab |
| Sign epistasis | 0 | 1 | -1 | 2 | AB has opposite sign to Ab or aB |
| Reciprocal sign epistasis | 0 | -1 | -1 | 2 | AB has opposite sign to Ab and aB |
Diploid organisms
Epistasis in diploid organisms is further complicated by the presence of two copies of each gene. Epistasis can occur between loci, but additionally, interactions can occur between the two copies of each locus in heterozygotes. For a two locus, two allele system, there are eight independent types of gene interaction.[27]
| Additive A locus | Additive B locus | Dominance A locus | Dominance B locus | ||||||||||||||||
| aa | aA | AA | aa | aA | AA | aa | aA | AA | aa | aA | AA | ||||||||
| bb | 1 | 0 | –1 | bb | 1 | 1 | 1 | bb | –1 | 1 | –1 | bb | –1 | –1 | –1 | ||||
| bB | 1 | 0 | –1 | bB | 0 | 0 | 0 | bB | –1 | 1 | –1 | bB | 1 | 1 | 1 | ||||
| BB | 1 | 0 | –1 | BB | –1 | –1 | –1 | BB | –1 | 1 | –1 | BB | –1 | –1 | –1 | ||||
| Additive by Additive Epistasis | Additive by Dominance Epistasis | Dominance by Additive Epistasis | Dominance by Dominance Epistasis | ||||||||||||||||
| aa | aA | AA | aa | aA | AA | aa | aA | AA | aa | aA | AA | ||||||||
| bb | 1 | 0 | –1 | bb | 1 | 0 | –1 | bb | 1 | –1 | 1 | bb | –1 | 1 | –1 | ||||
| bB | 0 | 0 | 0 | bB | –1 | 0 | 1 | bB | 0 | 0 | 0 | bB | 1 | –1 | 1 | ||||
| BB | –1 | 0 | 1 | BB | 1 | 0 | –1 | BB | –1 | 1 | –1 | BB | –1 | 1 | –1 | ||||
Genetic and molecular causes
Additivity
This can be the case when multiple genes act in parallel to achieve the same effect. For example, when an organism is in need of phosphorus, multiple enzymes that break down different phosphorylated components from the environment may act additively to increase the amount of phosphorus available to the organism. However, there inevitably comes a point where phosphorus is no longer the limiting factor for growth and reproduction and so further improvements in phosphorus metabolism have smaller or no effect (negative epistasis). Some sets of mutations within genes have also been specifically found to be additive.[28] It is now considered that strict additivity is the exception, rather than the rule, since most genes interact with hundreds or thousands of other genes.[18][19]
Epistasis between genes
Epistasis within the genomes of organisms occurs due to interactions between the genes within the genome. This interaction may be direct if the genes encode proteins that, for example, are separate components of a multi-component protein (such as the ribosome), inhibit each other's activity, or if the protein encoded by one gene modifies the other (such as by phosphorylation). Alternatively the interaction may be indirect, where the genes encode components of a metabolic pathway or network, developmental pathway, signalling pathway or transcription factor network. For example, the gene encoding the enzyme that synthesizes penicillin is of no use to a fungus without the enzymes that synthesize the necessary precursors in the metabolic pathway.
Epistasis within genes
Just as mutations in two separate genes can be non-additive if those genes interact, mutations in two codons within a gene can be non-additive. In genetics this is sometimes called intragenic suppression when one deleterious mutation can be compensated for by a second mutation within that gene. Analysis of bacteriophage T4 mutants that were altered in the rIIB cistron (gene) revealed that certain pairwise combinations of mutations could mutually suppress each other; that is the double mutants had a more nearly wild-type phenotype than either mutant alone.[29] The linear map order of the mutants was established using genetic recombination data, From these sources of information, the triplet nature of the genetic code was logically deduced for the first time in 1961, and other key features of the code were also inferred.[29]
Also intragenic suppression can occur when the amino acids within a protein interact. Due to the complexity of protein folding and activity, additive mutations are rare.
Proteins are held in their tertiary structure by a distributed, internal network of cooperative interactions (hydrophobic, polar and covalent).[30] Epistatic interactions occur whenever one mutation alters the local environment of another residue (either by directly contacting it, or by inducing changes in the protein structure).[31] For example, in a disulphide bridge, a single cysteine has no effect on protein stability until a second is present at the correct location at which point the two cysteines form a chemical bond which enhances the stability of the protein.[32] This would be observed as positive epistasis where the double-cysteine variant had a much higher stability than either of the single-cysteine variants. Conversely, when deleterious mutations are introduced, proteins often exhibit mutational robustness whereby as stabilising interactions are destroyed the protein still functions until it reaches some stability threshold at which point further destabilising mutations have large, detrimental effects as the protein can no longer fold. This leads to negative epistasis whereby mutations that have little effect alone have a large, deleterious effect together.[33][34]
In enzymes, the protein structure orients a few, key amino acids into precise geometries to form an active site to perform chemistry.[35] Since these active site networks frequently require the cooperation of multiple components, mutating any one of these components massively compromises activity, and so mutating a second component has a relatively minor effect on the already inactivated enzyme. For example, removing any member of the catalytic triad of many enzymes will reduce activity to levels low enough that the organism is no longer viable.[36][37][38]
Heterozygotic epistasis
Diploid organisms contain two copies of each gene. If these are different (heterozygous / heteroallelic), the two different copies of the allele may interact with each other to cause epistasis. This is sometimes called allelic complementation, or interallelic complementation. It may be caused by several mechanisms, for example transvection, where an enhancer from one allele acts in trans to activate transcription from the promoter of the second allele. Alternately, trans-splicing of two non-functional RNA molecules may produce a single, functional RNA.
Similarly, at the protein level, proteins that function as dimers may form a heterodimer composed of one protein from each alternate gene and may display different properties to the homodimer of one or both variants. Two bacteriophage T4 mutants defective at different locations in the same gene can undergo allelic complementation during a mixed infection.[39] That is, each mutant alone upon infection cannot produce viable progeny, but upon mixed infection with two complementing mutants, viable phage are formed. Intragenic complementation was demonstrated for several genes that encode structural proteins of the bacteriophage[39] indicating that such proteins function as dimers or even higher order multimers.[40]
Evolutionary consequences
Fitness landscapes and evolvability

In evolutionary genetics, the sign of epistasis is usually more significant than the magnitude of epistasis. This is because magnitude epistasis (positive and negative) simply affects how beneficial mutations are together, however sign epistasis affects whether mutation combinations are beneficial or deleterious.[10]
A fitness landscape is a representation of the fitness where all genotypes are arranged in 2D space and the fitness of each genotype is represented by height on a surface. It is frequently used as a visual metaphor for understanding evolution as the process of moving uphill from one genotype to the next, nearby, fitter genotype.[18]
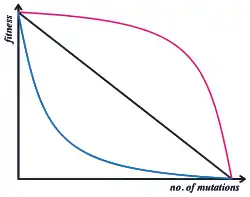
If all mutations are additive, they can be acquired in any order and still give a continuous uphill trajectory. The landscape is perfectly smooth, with only one peak (global maximum) and all sequences can evolve uphill to it by the accumulation of beneficial mutations in any order. Conversely, if mutations interact with one another by epistasis, the fitness landscape becomes rugged as the effect of a mutation depends on the genetic background of other mutations.[41] At its most extreme, interactions are so complex that the fitness is 'uncorrelated' with gene sequence and the topology of the landscape is random. This is referred to as a rugged fitness landscape and has profound implications for the evolutionary optimisation of organisms. If mutations are deleterious in one combination but beneficial in another, the fittest genotypes can only be accessed by accumulating mutations in one specific order. This makes it more likely that organisms will get stuck at local maxima in the fitness landscape having acquired mutations in the 'wrong' order.[34][42] For example, a variant of TEM1 β-lactamase with 5 mutations is able to cleave cefotaxime (a third generation antibiotic).[43] However, of the 120 possible pathways to this 5-mutant variant, only 7% are accessible to evolution as the remainder passed through fitness valleys where the combination of mutations reduces activity. In contrast, changes in environment (and therefore the shape of the fitness landscape) have been shown to provide escape from local maxima.[34] In this example, selection in changing antibiotic environments resulted in a "gateway mutation" which epistatically interacted in a positive manner with other mutations along an evolutionary pathway, effectively crossing a fitness valley. This gateway mutation alleviated the negative epistatic interactions of other individually beneficial mutations, allowing them to better function in concert. Complex environments or selections may therefore bypass local maxima found in models assuming simple positive selection.
High epistasis is usually considered a constraining factor on evolution, and improvements in a highly epistatic trait are considered to have lower evolvability. This is because, in any given genetic background, very few mutations will be beneficial, even though many mutations may need to occur to eventually improve the trait. The lack of a smooth landscape makes it harder for evolution to access fitness peaks. In highly rugged landscapes, fitness valleys block access to some genes, and even if ridges exist that allow access, these may be rare or prohibitively long.[44] Moreover, adaptation can move proteins into more precarious or rugged regions of the fitness landscape.[45] These shifting "fitness territories" may act to decelerate evolution and could represent tradeoffs for adaptive traits.
The frustration of adaptive evolution by rugged fitness landscapes was recognized as a potential force for the evolution of evolvability. Michael Conrad in 1972 was the first to propose a mechanism for the evolution of evolvability by noting that a mutation which smoothed the fitness landscape at other loci could facilitate the production of advantageous mutations and hitchhike along with them.[46][47] Rupert Riedl in 1975 proposed that new genes which produced the same phenotypic effects with a single mutation as other loci with reciprocal sign epistasis would be a new means to attain a phenotype otherwise too unlikely to occur by mutation.[48][49]
Rugged, epistatic fitness landscapes also affect the trajectories of evolution. When a mutation has a large number of epistatic effects, each accumulated mutation drastically changes the set of available beneficial mutations. Therefore, the evolutionary trajectory followed depends highly on which early mutations were accepted. Thus, repeats of evolution from the same starting point tend to diverge to different local maxima rather than converge on a single global maximum as they would in a smooth, additive landscape.[50][51]
Evolution of sex
Negative epistasis and sex are thought to be intimately correlated. Experimentally, this idea has been tested in using digital simulations of asexual and sexual populations. Over time, sexual populations move towards more negative epistasis, or the lowering of fitness by two interacting alleles. It is thought that negative epistasis allows individuals carrying the interacting deleterious mutations to be removed from the populations efficiently. This removes those alleles from the population, resulting in an overall more fit population. This hypothesis was proposed by Alexey Kondrashov, and is sometimes known as the deterministic mutation hypothesis[52] and has also been tested using artificial gene networks.[20]
However, the evidence for this hypothesis has not always been straightforward and the model proposed by Kondrashov has been criticized for assuming mutation parameters far from real world observations.[53] In addition, in those tests which used artificial gene networks, negative epistasis is only found in more densely connected networks,[20] whereas empirical evidence indicates that natural gene networks are sparsely connected,[54] and theory shows that selection for robustness will favor more sparsely connected and minimally complex networks.[54]
Methods and model systems
Regression analysis
Quantitative genetics focuses on genetic variance due to genetic interactions. Any two locus interactions at a particular gene frequency can be decomposed into eight independent genetic effects using a weighted regression. In this regression, the observed two locus genetic effects are treated as dependent variables and the "pure" genetic effects are used as the independent variables. Because the regression is weighted, the partitioning among the variance components will change as a function of gene frequency. By analogy it is possible to expand this system to three or more loci, or to cytonuclear interactions[55]
Double mutant cycles
When assaying epistasis within a gene, site-directed mutagenesis can be used to generate the different genes, and their protein products can be assayed (e.g. for stability or catalytic activity). This is sometimes called a double mutant cycle and involves producing and assaying the wild type protein, the two single mutants and the double mutant. Epistasis is measured as the difference between the effects of the mutations together versus the sum of their individual effects.[56] This can be expressed as a free energy of interaction. The same methodology can be used to investigate the interactions between larger sets of mutations but all combinations have to be produced and assayed. For example, there are 120 different combinations of 5 mutations, some or all of which may show epistasis...
Computational prediction
Numerous computational methods have been developed for the detection and characterization of epistasis. Many of these rely on machine learning to detect non-additive effects that might be missed by statistical approaches such as linear regression.[57] For example, multifactor dimensionality reduction (MDR) was designed specifically for nonparametric and model-free detection of combinations of genetic variants that are predictive of a phenotype such as disease status in human populations.[58][59] Several of these approaches have been broadly reviewed in the literature.[60] Even more recently, methods that utilize insights from theoretical computer science (the Hadamard transform[61] and compressed sensing[62][63]) or maximum-likelihood inference[64] were shown to distinguish epistatic effects from overall non-linearity in genotype–phenotype map structure,[65] while others used patient survival analysis to identify non-linearity.[66]
See also
References
- Neil A. Campbell, Jane B. Reece: Biologie. Spektrum-Verlag Heidelberg-Berlin 2003, ISBN 3-8274-1352-4, page 306
- Gros PA, Le Nagard H, Tenaillon O (May 2009). "The Evolution of Epistasis and its Links with Genetic Robustness, Complexity and Drift in a Phenotypic Model of Adaptation". Genetics. 182 (1): 277–93. doi:10.1534/genetics.108.099127. PMC 2674823. PMID 19279327.
- Rieger R, Michaelis A, Green MM (1968), A Glossary of Genetics and Cytogenetics: Classical and Molecular, New York: Springer-Verlag, ISBN 978-0-387-07668-3
- Richmond, Marsha L. (2001). "Women in the Early History of Genetics: William Bateson and the Newnham College Mendelians, 1900–1910". Isis. The History of Science Society. 92 (1): 55–90. doi:10.1086/385040. JSTOR 237327. PMID 11441497. S2CID 29790111.
- Szendro IG, Martijn F S, Franke J, Krug J, de Visser J, Arjan GM (16 January 2013). "Quantitative analyses of empirical fitness landscapes". Journal of Statistical Mechanics: Theory and Experiment. 2013 (1): P01005. arXiv:1202.4378. Bibcode:2013JSMTE..01..005S. doi:10.1088/1742-5468/2013/01/P01005. S2CID 14356766.
- Edlund JA, Adami C (Spring 2004). "Evolution of robustness in digital organisms". Artificial Life. 10 (2): 167–79. CiteSeerX 10.1.1.556.2318. doi:10.1162/106454604773563595. PMID 15107229. S2CID 13371337.
- Chattopadhyay S (Spring 2019). "Genome-wide interaction and pathway-based identification of key regulators in multiple myeloma". Communications Biology. 4 (2): 89–96. doi:10.1038/s42003-019-0329-2. PMC 6399257. PMID 30854481.
- Ayati, Marzieh; Koyutürk, Mehmet (2014). "Prioritization of genomic locus pairs for testing epistasis". Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics. pp. 240–248. CiteSeerX 10.1.1.715.1549. doi:10.1145/2649387.2649449. ISBN 978-1-4503-2894-4. S2CID 17343957.
- Piriyapongsa J, Ngamphiw C, Intarapanich A, Kulawonganunchai S, Assawamakin A, Bootchai C, Shaw PJ, Tongsima S (2012-12-13). "iLOCi: a SNP interaction prioritization technique for detecting epistasis in genome-wide association studies". BMC Genomics. 13 (Suppl 7): S2. doi:10.1186/1471-2164-13-S7-S2. PMC 3521387. PMID 23281813.
- Phillips PC (November 2008). "Epistasis--the essential role of gene interactions in the structure and evolution of genetic systems". Nature Reviews. Genetics. 9 (11): 855–67. doi:10.1038/nrg2452. PMC 2689140. PMID 18852697.
- Domingo E, Sheldon J, Perales C (June 2012). "Viral quasispecies evolution". Microbiology and Molecular Biology Reviews. 76 (2): 159–216. doi:10.1128/mmbr.05023-11. PMC 3372249. PMID 22688811.
- Tokuriki N, Tawfik DS (October 2009). "Stability effects of mutations and protein evolvability". Current Opinion in Structural Biology. 19 (5): 596–604. doi:10.1016/j.sbi.2009.08.003. PMID 19765975.
- He X, Qian W, Wang Z, Li Y, Zhang J (March 2010). "Prevalent positive epistasis in Escherichia coli and Saccharomyces cerevisiae metabolic networks". Nature Genetics. 42 (3): 272–6. doi:10.1038/ng.524. PMC 2837480. PMID 20101242.
- Ridley M (2004). Evolution (3rd ed.). Blackwell Publishing.
- Charlesworth B, Charlesworth D (2010). Elements of Evolutionary Genetics. Roberts and Company Publishers.
- Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW (September 2007). "Crystal structure of an ancient protein: evolution by conformational epistasis". Science. 317 (5844): 1544–8. Bibcode:2007Sci...317.1544O. doi:10.1126/science.1142819. PMC 2519897. PMID 17702911.
- Cordell HJ (October 2002). "Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans". Human Molecular Genetics. 11 (20): 2463–8. CiteSeerX 10.1.1.719.4634. doi:10.1093/hmg/11.20.2463. PMID 12351582.
- Kauffman, Stuart A. (1993). The Origins of Order: Self-organization and Selection in Evolution. Oxford University Press. ISBN 978-0-19-507951-7.
- Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (May 2012). "Engineering the third wave of biocatalysis". Nature. 485 (7397): 185–94. Bibcode:2012Natur.485..185B. doi:10.1038/nature11117. PMID 22575958. S2CID 4379415.
- Azevedo RB, Lohaus R, Srinivasan S, Dang KK, Burch CL (March 2006). "Sexual reproduction selects for robustness and negative epistasis in artificial gene networks". Nature. 440 (7080): 87–90. Bibcode:2006Natur.440...87A. doi:10.1038/nature04488. PMID 16511495. S2CID 4415072.
- Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ (November 2004). "Evidence for positive epistasis in HIV-1". Science. 306 (5701): 1547–50. Bibcode:2004Sci...306.1547B. doi:10.1126/science.1101786. PMID 15567861. S2CID 45784964.
- Weinreich DM, Watson RA, Chao L (June 2005). "Perspective: Sign epistasis and genetic constraint on evolutionary trajectories". Evolution; International Journal of Organic Evolution. 59 (6): 1165–74. doi:10.1111/j.0014-3820.2005.tb01768.x. JSTOR 3448895. PMID 16050094.
- Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ (January 2007). "Empirical fitness landscapes reveal accessible evolutionary paths". Nature. 445 (7126): 383–6. Bibcode:2007Natur.445..383P. doi:10.1038/nature05451. PMID 17251971. S2CID 4415468.
- Kaznatcheev, Artem (1 May 2019). "Computational Complexity as an Ultimate Constraint on Evolution". Genetics. 212 (1): 245–265. doi:10.1534/genetics.119.302000. PMC 6499524. PMID 30833289.
- Floor, E. (1970). "Interaction of morphogenetic genes of bacteriophage T4". Journal of Molecular Biology. 47 (3): 293–306. doi:10.1016/0022-2836(70)90303-7. PMID 4907266.
- French-Mischo S (July 2002). "Synthetic Lethal Mutations". Department of Microbiology, University of Illinois, Urbana. Archived from the original on 2016-08-23. Retrieved 2017-08-03.
- Kempthorne O (1969). An introduction to genetic statistics. Iowa State University Press. ISBN 978-0-8138-2375-1.
- Lunzer M, Miller SP, Felsheim R, Dean AM (October 2005). "The biochemical architecture of an ancient adaptive landscape". Science. 310 (5747): 499–501. Bibcode:2005Sci...310..499L. doi:10.1126/science.1115649. PMID 16239478. S2CID 28379541.
- Crick, F. H.; Barnett, L-; Brenner, S.; Watts-Tobin, R. J. (1961). "General nature of the genetic code for proteins". Nature. 192 (4809): 1227–1232. Bibcode:1961Natur.192.1227C. doi:10.1038/1921227a0. PMID 13882203. S2CID 4276146.
- Shakhnovich BE, Deeds E, Delisi C, Shakhnovich E (March 2005). "Protein structure and evolutionary history determine sequence space topology". Genome Research. 15 (3): 385–92. arXiv:q-bio/0404040. doi:10.1101/gr.3133605. PMC 551565. PMID 15741509.
- Harms MJ, Thornton JW (August 2013). "Evolutionary biochemistry: revealing the historical and physical causes of protein properties". Nature Reviews. Genetics. 14 (8): 559–71. doi:10.1038/nrg3540. PMC 4418793. PMID 23864121.
- Witt D (2008). "Recent developments in disulfide bond formation". Synthesis. 2008 (16): 2491–2509. doi:10.1055/s-2008-1067188.
- Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS (December 2006). "Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein". Nature. 444 (7121): 929–32. Bibcode:2006Natur.444..929B. doi:10.1038/nature05385. PMID 17122770. S2CID 4416275.
- Steinberg B, Ostermeier M (January 2016). "Environmental changes bridge evolutionary valleys". Science Advances. 2 (1): e1500921. Bibcode:2016SciA....2E0921S. doi:10.1126/sciadv.1500921. PMC 4737206. PMID 26844293.
- Halabi N, Rivoire O, Leibler S, Ranganathan R (August 2009). "Protein sectors: evolutionary units of three-dimensional structure". Cell. 138 (4): 774–86. doi:10.1016/j.cell.2009.07.038. PMC 3210731. PMID 19703402.
- Neet KE, Koshland DE (November 1966). "The conversion of serine at the active site of subtilisin to cysteine: a "chemical mutation"". Proceedings of the National Academy of Sciences of the United States of America. 56 (5): 1606–11. Bibcode:1966PNAS...56.1606N. doi:10.1073/pnas.56.5.1606. PMC 220044. PMID 5230319.
- Beveridge AJ (July 1996). "A theoretical study of the active sites of papain and S195C rat trypsin: implications for the low reactivity of mutant serine proteinases". Protein Science. 5 (7): 1355–65. doi:10.1002/pro.5560050714. PMC 2143470. PMID 8819168.
- Sigal IS, Harwood BG, Arentzen R (December 1982). "Thiol-beta-lactamase: replacement of the active-site serine of RTEM beta-lactamase by a cysteine residue". Proceedings of the National Academy of Sciences of the United States of America. 79 (23): 7157–60. Bibcode:1982PNAS...79.7157S. doi:10.1073/pnas.79.23.7157. PMC 347297. PMID 6818541.
- Bernstein, H.; Edgar, R. S.; Denhardt, G. H. (1965). "Intragenic Complementation Among Temperature Sensitive Mutants of Bacteriophage T4D". Genetics. 51 (6): 987–1002. doi:10.1093/genetics/51.6.987. PMC 1210828. PMID 14337770.
- CRICK FH; ORGEL LE (1964). "The Theory of Inter-Allelic Complementation". Journal of Molecular Biology. 8: 161–165. doi:10.1016/s0022-2836(64)80156-x. PMID 14149958.
- Poelwijk FJ, Tănase-Nicola S, Kiviet DJ, Tans SJ (March 2011). "Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes" (PDF). Journal of Theoretical Biology. 272 (1): 141–4. Bibcode:2011JThBi.272..141P. doi:10.1016/j.jtbi.2010.12.015. PMID 21167837.
- Reetz MT, Sanchis J (September 2008). "Constructing and analyzing the fitness landscape of an experimental evolutionary process". ChemBioChem. 9 (14): 2260–7. doi:10.1002/cbic.200800371. PMID 18712749. S2CID 23771783.
- Weinreich DM, Delaney NF, Depristo MA, Hartl DL (April 2006). "Darwinian evolution can follow only very few mutational paths to fitter proteins". Science. 312 (5770): 111–114. Bibcode:2006Sci...312..111W. doi:10.1126/science.1123539. PMID 16601193. S2CID 21186834.
- Gong LI, Suchard MA, Bloom JD (May 2013). "Stability-mediated epistasis constrains the evolution of an influenza protein". eLife. 2: e00631. doi:10.7554/eLife.00631. PMC 3654441. PMID 23682315.
- Steinberg B, Ostermeier M (July 2016). "Shifting Fitness and Epistatic Landscapes Reflect Trade-offs along an Evolutionary Pathway". Journal of Molecular Biology. 428 (13): 2730–43. doi:10.1016/j.jmb.2016.04.033. PMID 27173379.
- Conrad M (1972). "The importance of molecular hierarchy in information processing". Towards a Theoretical Biology. Edinburgh University Press Edinburgh (4): 222.
- Conrad, Michael (1979). "Bootstrapping on the adaptive landscape". BioSystems. 11 (2–3): 167–182. doi:10.1016/0303-2647(79)90009-1. hdl:2027.42/23514. PMID 497367.
- Riedl, Rupert J. (1975). Die Ordnung des Lebendigen: Systembedingungen der Evolution. Hamburg and Berlin: Parey.
- Riedl, Rupert J. (1977). "A systems-analytical approach to macroevolutionary phenomena". Quarterly Review of Biology. 52 (4): 351–370. doi:10.1086/410123. PMID 343152. S2CID 25465466.
- Lobkovsky AE, Wolf YI, Koonin EV (December 2011). "Predictability of evolutionary trajectories in fitness landscapes". PLOS Computational Biology. 7 (12): e1002302. arXiv:1108.3590. Bibcode:2011PLSCB...7E2302L. doi:10.1371/journal.pcbi.1002302. PMC 3240586. PMID 22194675.
- Bridgham JT, Ortlund EA, Thornton JW (September 2009). "An epistatic ratchet constrains the direction of glucocorticoid receptor evolution". Nature. 461 (7263): 515–9. Bibcode:2009Natur.461..515B. doi:10.1038/nature08249. PMC 6141187. PMID 19779450.
- Kondrashov AS (December 1988). "Deleterious mutations and the evolution of sexual reproduction". Nature. 336 (6198): 435–40. Bibcode:1988Natur.336..435K. doi:10.1038/336435a0. PMID 3057385. S2CID 4233528.
- MacCarthy T, Bergman A (July 2007). "Coevolution of robustness, epistasis, and recombination favors asexual reproduction". Proceedings of the National Academy of Sciences of the United States of America. 104 (31): 12801–6. Bibcode:2007PNAS..10412801M. doi:10.1073/pnas.0705455104. PMC 1931480. PMID 17646644.
- Leclerc RD (August 2008). "Survival of the sparsest: robust gene networks are parsimonious". Molecular Systems Biology. 4 (213): 213. doi:10.1038/msb.2008.52. PMC 2538912. PMID 18682703.
- Wade MJ, Goodnight CJ (April 2006). "Cyto-nuclear epistasis: two-locus random genetic drift in hermaphroditic and dioecious species". Evolution; International Journal of Organic Evolution. 60 (4): 643–59. doi:10.1554/05-019.1. PMID 16739448. S2CID 41900960.
- Horovitz A (1996). "Double-mutant cycles: a powerful tool for analyzing protein structure and function". Folding & Design. 1 (6): R121–6. doi:10.1016/s1359-0278(96)00056-9. PMID 9080186.
- Chicco D, Faultless T (2021-03-18). "Brief Survey on Machine Learning in Epistasis". Epistasis. Methods in Molecular Biology. Vol. 2212. pp. 169–179. doi:10.1007/978-1-0716-0947-7_11. ISBN 978-1-0716-0947-7. PMID 33733356. S2CID 232303194.
- Moore JH, Andrews PC (2015-01-01). "Epistasis Analysis Using Multifactor Dimensionality Reduction". Epistasis. Methods in Molecular Biology. Vol. 1253. pp. 301–14. doi:10.1007/978-1-4939-2155-3_16. ISBN 978-1-4939-2154-6. PMID 25403539.
- Moore JH, Williams SM, eds. (2015). Epistasis: Methods and Protocols. Methods in Molecular Biology. Vol. 1253. Springer. doi:10.1007/978-1-4939-2155-3. ISBN 978-1-4939-2154-6. S2CID 241378477.
- Cordell HJ (June 2009). "Detecting gene-gene interactions that underlie human diseases". Nature Reviews. Genetics. 10 (6): 392–404. doi:10.1038/nrg2579. PMC 2872761. PMID 19434077.
- Weinreich DM, Lan Y, Wylie CS, Heckendorn RB (December 2013). "Should evolutionary geneticists worry about higher-order epistasis?". Current Opinion in Genetics & Development. Genetics of system biology. 23 (6): 700–7. doi:10.1016/j.gde.2013.10.007. PMC 4313208. PMID 24290990.
- Poelwijk FJ, Krishna V, Ranganathan R (June 2016). "The Context-Dependence of Mutations: A Linkage of Formalisms". PLOS Computational Biology. 12 (6): e1004771. arXiv:1502.00726. Bibcode:2016PLSCB..12E4771P. doi:10.1371/journal.pcbi.1004771. PMC 4919011. PMID 27337695.
- Poelwijk FJ, Socolich, M, Ranganathan R (September 2019). "Learning the pattern of epistasis linking genotype and phenotype in a protein". Nature Communications. 10 (1): 4213. Bibcode:2019NatCo..10.4213P. doi:10.1038/s41467-019-12130-8. PMC 6746860. PMID 31527666.
- Otwinowski J, McCandlish DM, Plotkin JB (August 2018). "Inferring the shape of global epistasis". Proceedings of the National Academy of Sciences of the United States of America. 115 (32): E7550–E7558. Bibcode:2018PNAS..115E7550O. doi:10.1073/pnas.1804015115. PMC 6094095. PMID 30037990.
- Sailer ZR, Harms MJ (March 2017). "Detecting High-Order Epistasis in Nonlinear Genotype-Phenotype Maps". Genetics. 205 (3): 1079–1088. doi:10.1534/genetics.116.195214. PMC 5340324. PMID 28100592.
- Magen, A (2019). "Beyond Synthetic Lethality: Charting the Landscape of Pairwise Gene Expression States Associated with Survival in Cancer". Cell Reports. 28 (4): P938–948.E6. doi:10.1016/j.celrep.2019.06.067. PMC 8261641. PMID 31340155.
External links
- INTERSNP - a software for genome-wide interaction analysis (GWIA) of case-control and case-only SNP data, including analysis of quantitative traits.
- Science Aid: Epistasis High school (GCSE, Alevel) resource.
- GeneticInteractions.org
- Epistasis.org