Transition state theory
In chemistry, transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.[1]
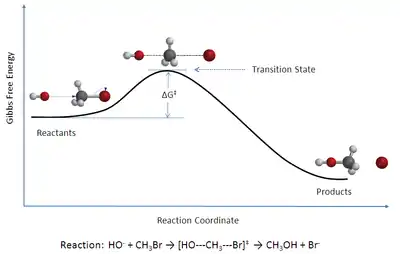
TST is used primarily to understand qualitatively how chemical reactions take place. TST has been less successful in its original goal of calculating absolute reaction rate constants because the calculation of absolute reaction rates requires precise knowledge of potential energy surfaces,[2] but it has been successful in calculating the standard enthalpy of activation (ΔH‡, also written Δ‡Hɵ), the standard entropy of activation (ΔS‡ or Δ‡Sɵ), and the standard Gibbs energy of activation (ΔG‡ or Δ‡Gɵ) for a particular reaction if its rate constant has been experimentally determined. (The ‡ notation refers to the value of interest at the transition state; ΔH‡ is the difference between the enthalpy of the transition state and that of the reactants.)
This theory was developed simultaneously in 1935 by Henry Eyring, then at Princeton University, and by Meredith Gwynne Evans and Michael Polanyi of the University of Manchester.[3][4] TST is also referred to as "activated-complex theory", "absolute-rate theory", and "theory of absolute reaction rates".[5]
Before the development of TST, the Arrhenius rate law was widely used to determine energies for the reaction barrier. The Arrhenius equation derives from empirical observations and ignores any mechanistic considerations, such as whether one or more reactive intermediates are involved in the conversion of a reactant to a product.[6] Therefore, further development was necessary to understand the two parameters associated with this law, the pre-exponential factor (A) and the activation energy (Ea). TST, which led to the Eyring equation, successfully addresses these two issues; however, 46 years elapsed between the publication of the Arrhenius rate law, in 1889, and the Eyring equation derived from TST, in 1935. During that period, many scientists and researchers contributed significantly to the development of the theory.
Theory
The basic ideas behind transition state theory are as follows:
- Rates of reaction can be studied by examining activated complexes near the saddle point of a potential energy surface. The details of how these complexes are formed are not important. The saddle point itself is called the transition state.
- The activated complexes are in a special equilibrium (quasi-equilibrium) with the reactant molecules.
- The activated complexes can convert into products, and kinetic theory can be used to calculate the rate of this conversion.
Development
In the development of TST, three approaches were taken as summarized below
Thermodynamic treatment
In 1884, Jacobus van 't Hoff proposed the Van 't Hoff equation describing the temperature dependence of the equilibrium constant for a reversible reaction:


where ΔU is the change in internal energy, K is the equilibrium constant of the reaction, R is the universal gas constant, and T is thermodynamic temperature. Based on experimental work, in 1889, Svante Arrhenius proposed a similar expression for the rate constant of a reaction, given as follows:

Integration of this expression leads to the Arrhenius equation

where k is the rate constant. A was referred to as the frequency factor (now called the pre-exponential coefficient), and Ea is regarded as the activation energy. By the early 20th century many had accepted the Arrhenius equation, but the physical interpretation of A and Ea remained vague. This led many researchers in chemical kinetics to offer different theories of how chemical reactions occurred in an attempt to relate A and Ea to the molecular dynamics directly responsible for chemical reactions.
In 1910, French chemist René Marcelin introduced the concept of standard Gibbs energy of activation. His relation can be written as

At about the same time as Marcelin was working on his formulation, Dutch chemists Philip Abraham Kohnstamm, Frans Eppo Cornelis Scheffer, and Wiedold Frans Brandsma introduced standard entropy of activation and the standard enthalpy of activation. They proposed the following rate constant equation

However, the nature of the constant was still unclear.
Kinetic-theory treatment
In early 1900, Max Trautz and William Lewis studied the rate of the reaction using collision theory, based on the kinetic theory of gases. Collision theory treats reacting molecules as hard spheres colliding with one another; this theory neglects entropy changes, since it assumes that the collision between molecules are completely elastic.
Lewis applied his treatment to the following reaction and obtained good agreement with experimental result.
2HI → H2 + I2
However, later when the same treatment was applied to other reactions, there were large discrepancies between theoretical and experimental results.
Statistical-mechanical treatment
Statistical mechanics played a significant role in the development of TST. However, the application of statistical mechanics to TST was developed very slowly given the fact that in mid-19th century, James Clerk Maxwell, Ludwig Boltzmann, and Leopold Pfaundler published several papers discussing reaction equilibrium and rates in terms of molecular motions and the statistical distribution of molecular speeds.
It was not until 1912 when the French chemist A. Berthoud used the Maxwell–Boltzmann distribution law to obtain an expression for the rate constant.

where a and b are constants related to energy terms.
Two years later, René Marcelin made an essential contribution by treating the progress of a chemical reaction as a motion of a point in phase space. He then applied Gibbs' statistical-mechanical procedures and obtained an expression similar to the one he had obtained earlier from thermodynamic consideration.
In 1915, another important contribution came from British physicist James Rice. Based on his statistical analysis, he concluded that the rate constant is proportional to the "critical increment". His ideas were further developed by Richard Chace Tolman. In 1919, Austrian physicist Karl Ferdinand Herzfeld applied statistical mechanics to the equilibrium constant and kinetic theory to the rate constant of the reverse reaction, k−1, for the reversible dissociation of a diatomic molecule.[7]
![{\displaystyle {\ce {AB <=>[k_1][k_{-1}] {A}+ {B}}}}](../I/8df558c371c7f125f5833608e30f847abe2601de.svg)
He obtained the following equation for the rate constant of the forward reaction[8]

where is the dissociation energy at absolute zero, kB is the Boltzmann constant, h is the Planck constant, T is thermodynamic temperature, is vibrational frequency of the bond. This expression is very important since it is the first time that the factor kBT/h, which is a critical component of TST, has appeared in a rate equation.


In 1920, the American chemist Richard Chace Tolman further developed Rice's idea of the critical increment. He concluded that critical increment (now referred to as activation energy) of a reaction is equal to the average energy of all molecules undergoing reaction minus the average energy of all reactant molecules.
Potential energy surfaces
The concept of potential energy surface was very important in the development of TST. The foundation of this concept was laid by René Marcelin in 1913. He theorized that the progress of a chemical reaction could be described as a point in a potential energy surface with coordinates in atomic momenta and distances.
In 1931, Henry Eyring and Michael Polanyi constructed a potential energy surface for the reaction below. This surface is a three-dimensional diagram based on quantum-mechanical principles as well as experimental data on vibrational frequencies and energies of dissociation.
H + H2 → H2 + H
A year after the Eyring and Polanyi construction, Hans Pelzer and Eugene Wigner made an important contribution by following the progress of a reaction on a potential energy surface. The importance of this work was that it was the first time that the concept of col or saddle point in the potential energy surface was discussed. They concluded that the rate of a reaction is determined by the motion of the system through that col.
It has been typically assumed that the rate-limiting or lowest saddle point is located on the same energy surface as the initial ground state. However, it was recently found that this could be incorrect for processes occurring in semiconductors and insulators, where an initial excited state could go through a saddle point lower than the one on the surface of the initial ground state.[9]
Kramers theory of reaction rates
By modeling reactions as Langevin motion along a one dimensional reaction coordinate, Hendrik Kramers was able to derive a relationship between the shape of the potential energy surface along the reaction coordinate and the transition rates of the system. The formulation relies on approximating the potential energy landscape as a series of harmonic wells. In a two state system, there will be three wells; a well for state A, an upside-down well representing the potential energy barrier, and a well for state B.
In the overdamped (or "diffusive") regime, the transition rate from state A to B is related to the resonant frequency of the wells via

where is the frequency of the well for state A, is the frequency of the barrier well, is the viscous damping, is the energy of the top of the barrier, is the energy of bottom of the well for state A, and is the temperature of the system times the Boltzmann constant.[10]






For general damping (overdamped or underdamped), there is a similar formula.[11]
Justification for the Eyring equation
One of the most important features introduced by Eyring, Polanyi and Evans was the notion that activated complexes are in quasi-equilibrium with the reactants. The rate is then directly proportional to the concentration of these complexes multiplied by the frequency (kBT/h) with which they are converted into products. Below, a non-rigorous plausibility argument is given for the functional form of the Eyring equation. However, the key statistical mechanical factor kBT/h will not be justified, and the argument presented below does not constitute a true "derivation" of the Eyring equation.[12]
Quasi-equilibrium assumption
Quasi-equilibrium is different from classical chemical equilibrium, but can be described using a similar thermodynamic treatment.[5] [13] Consider the reaction below
![{\displaystyle {\ce {{A}+{B}<=>{[AB]^{\ddagger }}->{P}}}}](../I/0b873373ba74f1671f87574af29e3a0a9ba9c63d.svg)
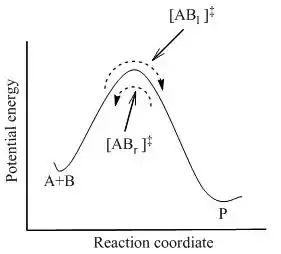
where complete equilibrium is achieved between all the species in the system including activated complexes, [AB]‡ . Using statistical mechanics, concentration of [AB]‡ can be calculated in terms of the concentration of A and B.
TST assumes that even when the reactants and products are not in equilibrium with each other, the activated complexes are in quasi-equilibrium with the reactants. As illustrated in Figure 2, at any instant of time, there are a few activated complexes, and some were reactant molecules in the immediate past, which are designated [ABl]‡ (since they are moving from left to right). The remainder of them were product molecules in the immediate past ([ABr]‡).
In TST, it is assumed that the flux of activated complexes in the two directions are independent of each other. That is, if all the product molecules were suddenly removed from the reaction system, the flow of [ABr]‡ stops, but there is still a flow from left to right. Hence, to be technically correct, the reactants are in equilibrium only with [ABl]‡, the activated complexes that were reactants in the immediate past.
Plausibility argument
The activated complexes do not follow a Boltzmann distribution of energies, but an "equilibrium constant" can still be derived from the distribution they do follow. The equilibrium constant K‡ for the quasi-equilibrium can be written as
- .
![{\displaystyle K^{\ddagger }={\frac {\ce {[AB]^{\ddagger }}}{\ce {[A][B]}}}}](../I/1465c535ba1f38d0eb964550468ee6c1f19aeb98.svg)
So, the chemical activity of the transition state AB‡ is
- .
![{\displaystyle [{\ce {AB}}]^{\ddagger }=K^{\ddagger }[{\ce {A}}][{\ce {B}}]}](../I/a67224b747342a2c608a96509499f845d991e801.svg)
Therefore, the rate equation for the production of product is
- ,
![{\displaystyle {\frac {d[{\ce {P}}]}{dt}}=k^{\ddagger }[{\ce {AB}}]^{\ddagger }=k^{\ddagger }K^{\ddagger }[{\ce {A}}][{\ce {B}}]=k[{\ce {A}}][{\ce {B}}]}](../I/618074b91e2c1707bac08640100ceed7c7da2214.svg)
where the rate constant k is given by
- .

Here, k‡ is directly proportional to the frequency of the vibrational mode responsible for converting the activated complex to the product; the frequency of this vibrational mode is . Every vibration does not necessarily lead to the formation of product, so a proportionality constant , referred to as the transmission coefficient, is introduced to account for this effect. So k‡ can be rewritten as

- .

For the equilibrium constant K‡ , statistical mechanics leads to a temperature dependent expression given as
- ().


Combining the new expressions for k‡ and K‡, a new rate constant expression can be written, which is given as
- .

Since, by definition, ΔG‡ = ΔH‡ –TΔS‡, the rate constant expression can be expanded, to give an alternative form of the Eyring equation:
- .

For correct dimensionality, the equation needs to have an extra factor of (c⊖)1–m for reactions that are not unimolecular:
- ,

where c⊖ is the standard concentration 1 mol⋅L–1 and m is the molecularity.[14]
Inferences from transition state theory and relationship with Arrhenius theory
The rate constant expression from transition state theory can be used to calculate the ΔG‡, ΔH‡, ΔS‡, and even ΔV‡ (the volume of activation) using experimental rate data. These so-called activation parameters give insight into the nature of a transition state, including energy content and degree of order, compared to the starting materials and has become a standard tool for elucidation of reaction mechanisms in physical organic chemistry. The free energy of activation, ΔG‡, is defined in transition state theory to be the energy such that holds. The parameters ΔH‡ and ΔS‡ can then be inferred by determining ΔG‡ = ΔH‡ – TΔS‡ at different temperatures.

Because the functional form of the Eyring and Arrhenius equations are similar, it is tempting to relate the activation parameters with the activation energy and pre-exponential factors of the Arrhenius treatment. However, the Arrhenius equation was derived from experimental data and models the macroscopic rate using only two parameters, irrespective of the number of transition states in a mechanism. In contrast, activation parameters can be found for every transition state of a multistep mechanism, at least in principle. Thus, although the enthalpy of activation, ΔH‡, is often equated with Arrhenius's activation energy Ea, they are not equivalent. For a condensed-phase (e.g., solution-phase) or unimolecular gas-phase reaction step, Ea = ΔH‡ + RT. For other gas-phase reactions, Ea = ΔH‡ + (1 − Δn‡)RT, where Δn‡ is the change in the number of molecules on forming the transition state.[15] (Thus, for a bimolecular gas-phase process, Ea = ΔH‡ + 2RT.)
The entropy of activation, ΔS‡, gives the extent to which transition state (including any solvent molecules involved in or perturbed by the reaction) is more disordered compared to the starting materials. It offers a concrete interpretation of the pre-exponential factor A in the Arrhenius equation; for a unimolecular, single-step process, the rough equivalence A = (kBT/h) exp(1 + ΔS‡/R) (or A = (kBT/h) exp(2 + ΔS‡/R) for bimolecular gas-phase reactions) holds. For a unimolecular process, a negative value indicates a more ordered, rigid transition state than the ground state, while a positive value reflects a transition state with looser bonds and/or greater conformational freedom. It is important to note that, for reasons of dimensionality, reactions that are bimolecular or higher have ΔS‡ values that depend on the standard state chosen (standard concentration, in particular). For most recent publications, 1 mol L–1 or 1 molar is chosen. Since this choice is a human construct, based on our definitions of units for molar quantity and volume, the magnitude and sign of ΔS‡ for a single reaction is meaningless by itself; only comparisons of the value with that of a reference reaction of "known" (or assumed) mechanism, made at the same standard state, is valid.[16]
The volume of activation is found by taking the partial derivative of ΔG‡ with respect to pressure (holding temperature constant): . It gives information regarding the size, and hence, degree of bonding at the transition state. An associative mechanism will likely have a negative volume of activation, while a dissociative mechanism will likely have a positive value.

Given the relationship between equilibrium constant and the forward and reverse rate constants, , the Eyring equation implies that

- .

Another implication of TST is the Curtin–Hammett principle: the product ratio of a kinetically-controlled reaction from R to two products A and B will reflect the difference in the energies of the respective transition states leading to product, assuming there is a single transition state to each one:
- ().
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=e^{-\Delta \Delta G^{\ddagger }/RT}}](../I/ac8276860d8cfb32f02aba8aba55acef114beb8b.svg)

(In the expression for ΔΔG‡ above, there is an extra term if A and B are formed from two different species SA and SB in equilibrium.)

For a thermodynamically-controlled reaction, every difference of RT ln 10 ≈ (1.987 × 10–3 kcal/mol K)(298 K)(2.303) ≈ 1.36 kcal/mol in the free energies of products A and B results in a factor of 10 in selectivity at room temperature (298 K), a principle known as the "1.36 rule":
- ().
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=10^{-\Delta G^{\circ }/(1.36\ \mathrm {kcal/mol} )}}](../I/91b01377882f2cca09a5e47e866e2f82b0a9a222.svg)

Analogously, every 1.36 kcal/mol difference in the free energy of activation results in a factor of 10 in selectivity for a kinetically-controlled process at room temperature:[17]
- ().
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=10^{-\Delta \Delta G^{\ddagger }/(1.36\ \mathrm {kcal/mol} )}}](../I/b17890458cfb78285761eed11143f5343072c979.svg)
Using the Eyring equation, there is a straightforward relationship between ΔG‡, first-order rate constants, and reaction half-life at a given temperature. At 298 K, a reaction with ΔG‡ = 23 kcal/mol has a rate constant of k ≈ 8.4 × 10–5 s–1 and a half life of t1/2 ≈ 2.3 hours, figures that are often rounded to k ~ 10–4 s–1 and t1/2 ~ 2 h. Thus, a free energy of activation of this magnitude corresponds to a typical reaction that proceeds to completion overnight at room temperature. For comparison, the cyclohexane chair flip has a ΔG‡ of about 11 kcal/mol with k ~ 105 s–1, making it a dynamic process that takes place rapidly (faster than the NMR timescale) at room temperature. At the other end of the scale, the cis/trans isomerization of 2-butene has a ΔG‡ of about 60 kcal/mol, corresponding to k ~ 10–31 s–1 at 298 K. This is a negligible rate: the half-life is 12 orders of magnitude longer than the age of the universe.[18]
Limitations
In general, TST has provided researchers with a conceptual foundation for understanding how chemical reactions take place. Even though the theory is widely applicable, it does have limitations. For example, when applied to each elementary step of a multi-step reaction, the theory assumes that each intermediate is long-lived enough to reach a Boltzmann distribution of energies before continuing to the next step. When the intermediates are very short-lived, TST fails.[19] In such cases, the momentum of the reaction trajectory from the reactants to the intermediate can carry forward to affect product selectivity. An example of such a reaction is the ring closure of cyclopentane biradicals generated from the gas-phase thermal decomposition of 2,3-diazabicyclo[2.2.1]hept-2-ene.[20][21]
Transition state theory is also based on the assumption that atomic nuclei behave according to classical mechanics.[22] It is assumed that unless atoms or molecules collide with enough energy to form the transition structure, then the reaction does not occur. However, according to quantum mechanics, for any barrier with a finite amount of energy, there is a possibility that particles can still tunnel across the barrier. With respect to chemical reactions this means that there is a chance that molecules will react, even if they do not collide with enough energy to overcome the energy barrier.[23] While this effect is negligible for reactions with large activation energies, it becomes an important phenomenon for reactions with relatively low energy barriers, since the tunneling probability increases with decreasing barrier height.
Transition state theory fails for some reactions at high temperature. The theory assumes the reaction system will pass over the lowest energy saddle point on the potential energy surface. While this description is consistent for reactions occurring at relatively low temperatures, at high temperatures, molecules populate higher energy vibrational modes; their motion becomes more complex and collisions may lead to transition states far away from the lowest energy saddle point. This deviation from transition state theory is observed even in the simple exchange reaction between diatomic hydrogen and a hydrogen radical.[24]
Given these limitations, several alternatives to transition state theory have been proposed. A brief discussion of these theories follows.
Generalized transition state theory
Any form of TST, such as microcanonical variational TST, canonical variational TST, and improved canonical variational TST, in which the transition state is not necessarily located at the saddle point, is referred to as generalized transition state theory.
Microcanonical variational TST
A fundamental flaw of transition state theory is that it counts any crossing of the transition state as a reaction from reactants to products or vice versa. In reality, a molecule may cross this "dividing surface" and turn around, or cross multiple times and only truly react once. As such, unadjusted TST is said to provide an upper bound for the rate coefficients. To correct for this, variational transition state theory varies the location of the dividing surface that defines a successful reaction in order to minimize the rate for each fixed energy. [25] The rate expressions obtained in this microcanonical treatment can be integrated over the energy, taking into account the statistical distribution over energy states, so as to give the canonical, or thermal rates.
Canonical variational TST
A development of transition state theory in which the position of the dividing surface is varied so as to minimize the rate constant at a given temperature.
Improved canonical variational TST
A modification of canonical variational transition state theory in which, for energies below the threshold energy, the position of the dividing surface is taken to be that of the microcanonical threshold energy. This forces the contributions to rate constants to be zero if they are below the threshold energy. A compromise dividing surface is then chosen so as to minimize the contributions to the rate constant made by reactants having higher energies.
Nonadiabatic TST
An expansion of TST to the reactions when two spin-states are involved simultaneously is called nonadiabatic transition state theory (NA-TST).
Semiclassical TST
Using vibrational perturbation theory, effects such as tunnelling and variational effects can be accounted for within the SCTST formalism.
Applications
Enzymatic reactions
Enzymes catalyze chemical reactions at rates that are astounding relative to uncatalyzed chemistry at the same reaction conditions. Each catalytic event requires a minimum of three or often more steps, all of which occur within the few milliseconds that characterize typical enzymatic reactions. According to transition state theory, the smallest fraction of the catalytic cycle is spent in the most important step, that of the transition state. The original proposals of absolute reaction rate theory for chemical reactions defined the transition state as a distinct species in the reaction coordinate that determined the absolute reaction rate. Soon thereafter, Linus Pauling proposed that the powerful catalytic action of enzymes could be explained by specific tight binding to the transition state species [26] Because reaction rate is proportional to the fraction of the reactant in the transition state complex, the enzyme was proposed to increase the concentration of the reactive species.
This proposal was formalized by Wolfenden and coworkers at University of North Carolina at Chapel Hill, who hypothesized that the rate increase imposed by enzymes is proportional to the affinity of the enzyme for the transition state structure relative to the Michaelis complex.[27] Because enzymes typically increase the non-catalyzed reaction rate by factors of 1010-1015, and Michaelis complexes often have dissociation constants in the range of 10−3-10−6 M, it is proposed that transition state complexes are bound with dissociation constants in the range of 10−14 -10−23 M. As substrate progresses from the Michaelis complex to product, chemistry occurs by enzyme-induced changes in electron distribution in the substrate. Enzymes alter the electronic structure by protonation, proton abstraction, electron transfer, geometric distortion, hydrophobic partitioning, and interaction with Lewis acids and bases. Analogs that resemble the transition state structures should therefore provide the most powerful noncovalent inhibitors known.
All chemical transformations pass through an unstable structure called the transition state, which is poised between the chemical structures of the substrates and products. The transition states for chemical reactions are proposed to have lifetimes near 10−13 seconds, on the order of the time of a single bond vibration. No physical or spectroscopic method is available to directly observe the structure of the transition state for enzymatic reactions, yet transition state structure is central to understanding enzyme catalysis since enzymes work by lowering the activation energy of a chemical transformation.
It is now accepted that enzymes function to stabilize transition states lying between reactants and products, and that they would therefore be expected to bind strongly any inhibitor that closely resembles such a transition state. Substrates and products often participate in several enzyme catalyzed reactions, whereas the transition state tends to be characteristic of one particular enzyme, so that such an inhibitor tends to be specific for that particular enzyme. The identification of numerous transition state inhibitors supports the transition state stabilization hypothesis for enzymatic catalysis.
Currently there is a large number of enzymes known to interact with transition state analogs, most of which have been designed with the intention of inhibiting the target enzyme. Examples include HIV-1 protease, racemases, β-lactamases, metalloproteinases, cyclooxygenases and many others.
Adsorption on surfaces and reactions on surfaces
Desorption as well as reactions on surfaces are straightforward to describe with transition state theory. Analysis of adsorption to a surface from a liquid phase can present a challenge due to lack of ability to assess the concentration of the solute near the surface. When full details are not available, it has been proposed that reacting species' concentrations should be normalized to the concentration of active surface sites, an approximation called the surface reactant equi-density approximation (SREA).[28]
Notes
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "transition state theory". doi:10.1351/goldbook.T06470
- Truhlar, D. G.; Garrett, B. C.; Klippenstein, S. J. (1996). "Current Status of Transition-State Theory". J. Phys. Chem. 100 (31): 12771–12800. doi:10.1021/jp953748q.
- Laidler, K.; King, C. (1983). "Development of transition-state theory". J. Phys. Chem. 87 (15): 2657. doi:10.1021/j100238a002.
- Laidler, K.; King, C. (1998). "A lifetime of transition-state theory". The Chemical Intelligencer. 4 (3): 39.
- Laidler, K. J. (1969). Theories of Chemical Reaction Rates. McGraw-Hill.
- Anslyn, E. V.; Dougherty, D. A. (2006). "Transition State Theory and Related Topics". Modern Physical Organic Chemistry. University Science Books. pp. 365–373. ISBN 1891389319.
- Herzfeld, K. E. (1919). "Zur Theorie der Reaktionsgeschwindigkeiten in Gasen". Annalen der Physik. 364 (15): 635–667. Bibcode:1919AnP...364..635H. doi:10.1002/andp.19193641504.
- Keith J. Laidler, Chemical Kinetics (3rd ed., Harper & Row 1987), p.88 ISBN 0-06-043862-2
- Luo, G.; Kuech, T. F.; Morgan, D. (2018). "Transition state redox during dynamical processes in semiconductors and insulators". NPG Asia Materials. 10 (4): 45–51. arXiv:1712.01686. Bibcode:2018npjAM..10...45L. doi:10.1038/s41427-018-0010-0. S2CID 67780897.
- Lindsay, Stuart (2010). Introduction to nanoscience. Oxford University Press. pp. 109–111.
- "23.2: Kramers' Theory". Chemistry LibreTexts. 2021-01-17. Retrieved 2023-06-11.
- For an introductory treatment of the statistical mechanics and an elementary derivation of the Eyring equation, see: Lowry and Richardson, Mechanism and Theory in Organic Chemistry, 3rd ed. (Harper & Row, 1987), pp. 248-253.
- Steinfeld, Jeffrey L.; Francisco, Joseph S.; Hase, William L. (1999). Chemical Kinetics and Dynamics (2nd ed.). Prentice-Hall. pp. 289–293. ISBN 0-13-737123-3.
- Laidler, Keith J. (1981). "Symbolism and terminology in chemical kinetics" (PDF). Pure and Applied Chemistry. IUPAC. 53: 753–771. Retrieved 9 August 2019.
See p.765, note m.
- Steinfeld, Jeffrey L.; Francisco, Joseph S.; Hase, William L. (1999). Chemical Kinetics and Dynamics (2nd ed.). Prentice-Hall. p. 302. ISBN 0-13-737123-3.
- Carpenter, Barry K. (1984). Determination of organic reaction mechanisms. New York: Wiley. ISBN 0471893692. OCLC 9894996.
- Lowry, Thomas H. (1987). Mechanism and theory in organic chemistry. Richardson, Kathleen Schueller. (3rd ed.). New York: Harper & Row. ISBN 0060440848. OCLC 14214254.
- Eliel, Ernest L. (Ernest Ludwig) (1994). Stereochemistry of organic compounds. Wilen, Samuel H., Mander, Lewis N. New York: Wiley. ISBN 0471016705. OCLC 27642721.
- Carpenter, Barry K. (31 December 1998). "Dynamic Behavior of Organic Reactive Intermediates". Angewandte Chemie International Edition. 37 (24): 3340–3350. doi:10.1002/(SICI)1521-3773(19981231)37:24<3340::AID-ANIE3340>3.0.CO;2-1. eISSN 1521-3773. ISSN 1433-7851.
- Dougherty, Dennis A.; Anslyn, Eric V. (2006). Modern Physical Organic Chemistry. Sausalito, CA, USA: University Science Books. p. 374. ISBN 9781891389313.
- Reyes, Mayra B.; Carpenter, Barry K. (1 October 2000). "Mechanism of Thermal Deazetization of 2,3-Diazabicyclo[2.2.1]hept-2-ene and Its Reaction Dynamics in Supercritical Fluids". Journal of the American Chemical Society. 122 (41): 10163–10176. doi:10.1021/ja0016809. eISSN 1520-5126. ISSN 0002-7863.
- Eyring, H. (1935). "The Activated Complex in Chemical Reactions". J. Chem. Phys. 3 (2): 107–115. Bibcode:1935JChPh...3..107E. doi:10.1063/1.1749604.
- Masel, R. (1996). Principles of Adsorption and Reactions on Solid Surfaces. New York: Wiley.
- Pineda, J. R.; Schwartz, S. D. (2006). "Protein dynamics and catalysis: The problems of transition state theory and the subtlety of dynamic control". Phil. Trans. R. Soc. B. 361 (1472): 1433–1438. doi:10.1098/rstb.2006.1877. PMC 1647311. PMID 16873129.
- Truhlar, D.; Garrett, B. (1984). "Variational Transition State Theory". Annu. Rev. Phys. Chem. 35: 159–189. Bibcode:1984ARPC...35..159T. doi:10.1146/annurev.pc.35.100184.001111.
- Pauling, L. (1948). "Chemical Achievement and Hope for the Future". American Scientist. 36: 50–58. PMID 18920436.
- Radzicka, A.; Wolfenden, R. (1995). "A proficient enzyme". Science. 267 (5194): 90–93. Bibcode:1995Sci...267...90R. doi:10.1126/science.7809611. PMID 7809611.
- Doyle, Peter J.; Savara, Aditya; Raiman, Stephen S. (2020). "Extracting meaningful standard enthalpies and entropies of activation for surface reactions from kinetic rates". Reaction Kinetics, Mechanisms and Catalysis. 129 (2): 551–581. doi:10.1007/s11144-020-01747-2. S2CID 211836011.
References
- Anslyn, Eric V.; Doughtery, Dennis A., Transition State Theory and Related Topics. In Modern Physical Organic Chemistry University Science Books: 2006; pp 365–373
- Cleland, W.W., Isotope Effects: Determination of Enzyme Transition State Structure. Methods in Enzymology 1995, 249, 341-373
- Laidler, K.; King, C., Development of transition-state theory. The Journal of Physical Chemistry 1983, 87, (15), 2657
- Laidler, K., A lifetime of transition-state theory. The Chemical Intelligencer 1998, 4, (3), 39
- Radzicka, A.; Woldenden, R., Transition State and Multisubstrate$Analog Inhibitors. Methods in Enzymology 1995, 249, 284-312
- Schramm, VL., Enzymatic Transition States and Transition State Analog Design. Annual Review of Biochemistry 1998, 67, 693-720
- Schramm, V.L., Enzymatic Transition State Theory and Transition State Analogue Design. Journal of Biological Chemistry 2007, 282, (39), 28297-28300