Trinucleotide repeat expansion
A trinucleotide repeat expansion, also known as a triplet repeat expansion, is the DNA mutation responsible for causing any type of disorder categorized as a trinucleotide repeat disorder. These are labelled in dynamical genetics as dynamic mutations.[1] Triplet expansion is caused by slippage during DNA replication, also known as "copy choice" DNA replication.[2] Due to the repetitive nature of the DNA sequence in these regions, 'loop out' structures may form during DNA replication while maintaining complementary base pairing between the parent strand and daughter strand being synthesized. If the loop out structure is formed from the sequence on the daughter strand this will result in an increase in the number of repeats. However, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. It appears that expansion of these repeats is more common than reduction. Generally, the larger the expansion the more likely they are to cause disease or increase the severity of disease. Other proposed mechanisms for expansion and reduction involve the interaction of RNA and DNA molecules.[3]
In addition to occurring during DNA replication, trinucleotide repeat expansion can also occur during DNA repair.[4] When a DNA trinucleotide repeat sequence is damaged, it may be repaired by processes such as homologous recombination, non-homologous end joining, mismatch repair or base excision repair. Each of these processes involves a DNA synthesis step in which strand slippage might occur leading to trinucleotide repeat expansion.[4]
The number of trinucleotide repeats appears to predict the progression, severity, and age of onset of Huntington's disease and similar trinucleotide repeat disorders.[5] Other human diseases in which triplet repeat expansion occurs are fragile X syndrome, several spinocerebellar ataxias, myotonic dystrophy and Friedreich's ataxia.[4]
History
The first documentation of anticipation in genetic disorders was in the 1800s. However, from the eyes of geneticists, this relationship was disregarded and attributed to ascertainment bias; because of this, it took almost 200 years for a link between onset of disease and trinucleotide repeats (TNR) to be acknowledged.[6]
The following findings of served as support for TNR's link to onset of disease; the detection of various repeats within these diseases demonstrated this relationship.
- In 1991, for fragile X syndrome, the fragile X mental retardation 1 (FMR-1) gene was found to contain a CGG expansion in its 5' untranslated region (UTR).[7] In addition, a CAG expansion was located in X-linked spinal and bulbar muscular atrophy (SBMA) sequences.[8] SMBA is the first "CAG / polygutamine" disease, which is a subcategory of repeat disorders.[9]
- In 1992, for myotonic dystrophy type 1 (DM1), CTG expansion was found in the myotonic dystrophy protein kinase (DMPK) 3' UTR.
- In 1993, for Huntington's disease (HD), a longer-than-usual CAG repeat with was found in the exon 1 coding sequence.[10]
Because of these discoveries, ideas involving anticipation in disease began to develop, and curiosity formed about how the causes could be related to TNRs.[6] After the breakthroughs, the four mechanisms for TNRs were determined, and more types of repeats were identified as well.[9] Repeat composition and location are used to determine the mechanism of a given expansion.[9] Onwards from 1995, it was also possible to observe the formation of hairpins in triplet repeats, which consisted of repeating CG pairs and a mismatch.[11]
During the decade after evidence that linked TNR to onset of disease was found, focus was placed on studying repeat length and dynamics on diseases, as well as investigating the mechanism behind parent-child disease inheritance.[6] Research has shown that there is a clear inverse relationship between the length of the repeats in parents and the age of disease onset in children; therefore, the lengths of TNRs are used to predict age of disease onset as well as outcome in clinical diagnosis.[12][13] In addition to this finding, another aspect of the diseases, the high variability of onset, was revealed.[6] Although the onset of HD could be predicted by examining TNR length inheritance, the onset could vary up to fourfold depending on the patient, leading to the possibility of existence of age-modifying factors for disease onset; there were notable efforts in this search.[14][15] Currently, CAG repeat length is considered the biggest onset age modifier for TNR diseases.[16][17]
Detection of TNRs was made difficult by limited technology and methods early on, and years passed before the development of sufficient ways to measure the repeats.[6] When PCR was first attempted in the detection of TNRs, multiple band artifacts were prevalent in the results, and this made recognition of TNRs troublesome; at the time, debate centered around whether disease was brought on by smaller amounts of short expansions or a small amount of long expansions.[6][18] Since then, accurate methods have been established over the years. Together, the following clinically necessary protocols have 99% accuracy in measuring TNRs.[6]
- Small-pool polymerase chain reaction (SP-PCR) allows for recognition of repeat changes, and originated from the growing necessity for a method that would provide more accurate measurement of TNRs. It has been useful for examining how TNRs vary between human and mice in blood, sperm, and somatic cells.[19][20]
- Southern blots are used to measure CGG repeats because CG-rich regions limit polymerase movement in PCR.[21][22]
Overall structure
These repetitive sequences lead to instability amongst the DNA strands after reaching a certain threshold number of repeats, which can result in DNA slippage during replication.[23] The most common and well-known triplet repeats are CAG, GCG, CTG, CGG, and GAA. During DNA replication, the strand being synthesized can misalign with its template strand due to the dynamic nature and flexibility of these triplet repeats.[24] This slippage allows for the strand to find a stable intermediate amongst itself through base pairing, forming a secondary structure other than a duplex.[24]
Location
In terms of location, these triplet repeats can be found in both coding and non-coding regions. CAG and GCN repeats, which lead to polyglutamine and polyalanine tracts respectively, are normally found in the coding regions.[25] At the 5' untranslated region, CGG and CAG repeats are found and responsible for fragile X syndrome and spinocerebellar ataxia 12.[25] At the 3' untranslated region, CTG repeats are found, while GAA repeats are located in the intron region. Other disease-causing repeats, but not triplet repeats, have been located in the promoter region.[25] Once the number of repeats exceeds normal levels, Triplet Repeat Expansions (TRE) become more likely and the number of triplet repeats can typically increase to around 100 in coding regions and up to thousands in non-coding regions.[25] This difference is due to overexpression of glutamine and alanine, which is selected against due to cell toxicity.[26]
Intermediates
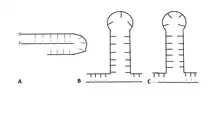
Depending on the sequence of the repeat, at least three intermediates with different secondary structures are known to form.[27] A CGG repeat will form a G-quadruplex due to Hoogsteen base pairing, while a GAA repeat forms a triplex due to negative supercoiling.[25][27] CAG, CTG, and CGG repeats form a hairpin. After the hairpin forms, the primer realigns with the 3' end of the newly synthesized strand and continues the synthesis, leading to triplet repeat expansion.[23] The structure of the hairpin is based on a stem and a loop that contains both Watson-Crick base pairs and mismatched pairs.[25] In CTG and CAG repeats, the number of nucleotides present in the loop depends on if the number of triplet repeats is odd or even.[28] An even number of repeats forms a tetraloop structure, while an odd number leads to the formation of a triloop.[28][29]
Instability
Threshold
In trinucleotide repeat expansion there is a certain threshold or maximum amount of repeats that can occur before a sequence becomes unstable. Once this threshold is reached the repeats will start to rapidly expand causing longer and longer expansions in future generations.[30] Once it hits this minimum allele size which is normally around 30-40 repeats, diseases and instability can be contracted, but if the number of repeats found within a sequence are below the threshold it will remain relatively stable.[30] There is still not enough research found to understand the molecular nature that causes thresholds but researchers are continuing to study that the possibility could lie with the formation of the secondary structure when these repeats occur. It was found that diseases associated with trinucleotide repeat expansions contained secondary structures with hairpins, triplexes, and slipped-strand duplexes.[30] These observations have led to the hypothesis that the threshold is determined by the number of repeats that must occur to stabilize the formation of these unwanted secondary structures, due to the fact that when these structures form there is an increased number of mutations[31] that will form in the sequence resulting in more trinucleotide expansion.
Parental influence
Research suggests that there is a direct, important correlation between the sex of the parent that transmits the mutation and the degree and phenotype of disorder in the child.,[32][33] The degree of repeat expansion and whether or not an expansion will occur has been directly linked to the sex of the transmitting parent in both non-coding and coding trinucleotide repeat disorders.[32] For example, research regarding the correlation between Huntington's Disease CAG trinucleotide repeat and parental transmission has found that there is a strong correlation between the two with differences in maternal and paternal transmission.[32] Maternal transmission has been observed to only consist of an increase in repeat units of 1 while the paternal transmission is typically anywhere from 3 to 9 extra repeats.[32] Paternal transmission is almost always responsible for large repeat transmission resulting in the early onset of Huntington's Disease while maternal transmission results in affected individuals experiencing symptom onset mirroring that of their mother.,[32][34] While this transmission of a trinucleotide repeat expansion is regarded to be a result of "meiotic instability", the degree to which meiosis plays a role in this process and the mechanism is not clear and numerous other processes are predicted to simultaneously play a role in this process.[32]
Mechanisms
Unequal homologous exchange
One proposed but highly unlikely mechanism that plays a role in trinucleotide expansion transmission occurs during meiotic or mitotic recombination.[32] It is suggested that during these processes it is possible for a homologous repeat misalignment, commonly known for causing alpha-globin locus deletions, causes the meiotic instability of a trinucleotide repeat expansion.[32] This process is unlikely to contribute to the transmission and presence of trinucleotide repeat expansions due to differences in expansion mechanisms.[32] Trinucleotide repeat expansions typically favor expansions of the CAG region but, in order for the unequal homologous exchange to be a plausible suggestion, these repeats would have to go through expansion and contraction events at the same time.[32] In addition, numerous diseases that result from transmitted trinucleotide repeat expansions, such as Fragile X syndrome, involve unstable trinucleotide repeats on the X chromosome that cannot be explained by meiotic recombination.[32] Research has shown that although unequal homologous recombination is unlikely to be the sole cause of transmitted trinucleotide repeat expansions, this homologous recombination likely plays a minor role in the length of some trinucleotide repeat expansions.[32]
DNA replication
DNA replication errors are predicted to be the main perpetrator of trinucleotide repeat expansion transmission in many predicted models due to the difficulty of Trinucleotide Repeat Expansion (TRE).[32] TREs have been shown to occur during DNA replication in both in vitro and in vivo studies, allowing for these long tracts of triplet repeats to assemble rapidly in different mechanisms that can result in either small scale or large scale expansions.[25]
Small scale expansions
These expansions can occur through either strand slippage or flap ligation.[25] Okazaki fragments are a key element of the proposed error in DNA replication.[32] It is suggested that the small size of Okazaki fragments, typically between 150 and 200 nucleotides long, makes them more likely to fall off or "slip" off the lagging strand, which creates room for trinucleotide repeats to attach to the lagging strand copy.[32] In addition to this possibility of trinucleotide repeat expansion changes occurring due to slippage of Okazaki fragments, the ability of CG-rich trinucleotide repeat expansion sequences to form a special hairpin, toroid, and triplex DNA structures contributes to this model, suggesting error occurs during DNA replication.[32] Hairpin structures can form as a result of the freedom of the lagging strand during DNA replication and are typically observed to form in extremely long trinucleotide repeat sequences.[32] Research has found that this hairpin formation depends on the orientation of the trinucleotide repeats within each CAG/CTG trinucleotide strand.[32] Strands that have duplex formation by CTG repeats in the leading strand are observed to result in extra repeats, while those without CTG repeats in the leading strand result in repeat deletions.[32] These intermediates can pause activity of the replication fork based on their interaction with DNA polymerases through strand slippage.[25] Contractions occur when the replication fork skips over the intermediate on the Okazaki fragment. Expansions occur when the fork reverses and restarts, which forms a chicken-foot structure.[25] This structure results in the unstable intermediate forming on the nascent leading strand, leading to further TRE.[25] Furthermore, this intermediate can avoid mismatch repair due to its affinity for the MSH-2-MSH3 complex, which stabilizes the hairpin instead of repairing it.[25][27] In non-dividing cells, a process called flap-ligation can be responsible for TRE.[25][27] 8-oxo-guanine DNA glycosylase removes a guanine and forms a nick in the sequence.[25] The coding strand then forms a flap due to displacement, which prevents removal by an endonuclease. When the repair process finishes for either mechanism, the length of the expansion is equivalent to the number of triplet repeats involved in the formation of the hairpin intermediate.[25]
Large scale expansions
Two mechanisms have been proposed for large scale repeats: template switching and break-induced replication.[27]
Template switching, a mechanism for large scale GAA repeats that can double the number of triplet repeats, has been proposed.[27] GAA repeats expand when their repeat length is greater than the Okazaki fragment's length.[27] These repeats are involved in the stalling of the replication fork as these repeats form a triplex when the 5' flap of TTC repeats fold back.[27] Okazaki fragment synthesis continues when the template is switched to the nascent leading strand.[27] The Okazaki fragment eventually ligates back to the 5' flap, which results in TRE.[27]
A different mechanism, based on break-induced replication, has been proposed for large scale CAG repeats and can also occur in non-dividing cells.[27] At first, this mechanism follows the same process as the small scale strand slippage mechanism until replication fork reversal.[27] An endonuclease then cleaves the chicken-foot structure, which results in a one-ended double strand break.[27] The CAG repeat of this broken daughter strand forms a hairpin and invades the CAG strand on the sister chromatid, which results in expansion of this repeat in a migrating D-loop DNA synthesis.[27] This synthesis continues until it reaches the replication fork and is cleaved, which results in an expanded sister chromatid.[27]
Disorders
Background
Fragile X syndrome is the second most common form of intellectual disability affecting 1 in 2,000-4,000 women and 1 in 4,000-8,000 men, women being twice as likely to inherit this disability due to their XX chromosomes.[35] This disability arises from a mutation at the end of the X chromosome in the FMR1 gene (Fragile X Mental Retardation Gene) which produces a protein essential for brain development called FMRP.[35] Individuals with Fragile X syndrome experience a variety of symptoms at varying degrees that depend on gender and mutation degree such as attention deficit disorders, irritability, stimuli sensitivity, various anxiety disorders, depression, and/or aggressive behavior.[35] Some treatments for these symptoms seen in individuals with Fragile X syndrome include SSRI's, antipsychotic medications, stimulants, folic acid, and mood stabilizers.[35]
Genetic causation
Sizable expansions of a CGG trinucleotide element are the singular cause of the male genetic disorder called Fragile X Syndrome. In males without Fragile X Syndrome, the CGG repeat number ranges from 53 to 200 while those affected have greater than 200 repeats of this trinucleotide sequence located at the end of the X chromosome on band Xq28.3.1.[36] Carriers that have repeats falling within the 53 to 200 repeat range are said to have "premutation alleles", as the alleles within this range approach 200, the likelihood of expansion to a full mutation increases, and the mRNA levels are elevated five-fold.[36] Research has shown that individuals with premutation alleles in the range of 59-69 repeats have about a 30% risk of developing full mutation and compared to those in the high range of ≥ 90 repeats.[37] Fragile X syndrome carriers (those that fall within the premutation range) typically have unmethylated alleles, normal phenotype, and normal levels of FMR1 mRNA and FMRP protein.[36] Fragile X Syndrome men possess alleles in the full mutation range (>200 repeats) with FMRP protein levels much lower than normal and experience hypermethylation of the promoter region of the FMR1 gene.[36] Some men with alleles in the full mutation range experience partial or no methylation which results in only slightly abnormal phenotypes due to only slight down-regulation of FMR1 gene transcription.[36] Unmethylated and partially methylated alleles in the mutation range experience increased and normal levels of FMR1 mRNA when compared to normal controls.[36] In contrast, when unmethylated alleles reach a repeat number of approximately 300, the transcription levels are relatively unaffected and operate at normal levels; the transcription levels of repeats greater than 300 is currently unknown.[36]
Promoter silencing
The CGG trinucleotide repeat expansion is present within the FMR1 mRNA and its interactions are responsible for promoter silencing.[36] The CGG trinucleotide expansion resides within the 5' untranslated region of the mRNA, which undergoes hybridization to form a complementary CGG repeat portion.[36] The binding of this genomic repeat to the mRNA results in silencing of the promoter.[36] Beyond this point, the mechanism of promoter silencing is unknown and still being further investigated.[36]
Background
Huntington's disease (HD) is a dominantly, paternally transmitted neurological disorder that affects 1 in 15,000-20,000 people in many Western Populations.[38] HD involves the basal ganglia and the cerebral cortex and manifests as symptoms such as cognitive, motor, and/or psychiatric impairment.[38]
Causation
This autosomal dominant disorder results from the expansions of a trinucleotide repeat which involves CAG in exon 1 of the IT15 gene.[39] The majority of all juvenile HD cases stem from the transmission of a high CAG trinucleotide repeat number that is a result of paternal gametogenesis.[40] While an individual without HD has a number of CAG repeats that fall within a range between 9 and 37, an individual with HD has CAG is typically found to have repeats in a range between 37 and 102.[39] Research has shown an inverse relationship between the number of trinucleotide repeats and age of onset, however, no relationship between trinucleotide repeat numbers and rate of HD progression and/or effected individual's body weight has been observed.[39] Severity of functional decline has been found to be similar across a wide range of individuals with varying numbers of CAG repeats and differing ages of onset, therefore, it is suggested that the rate of disease progression is also linked to factors other than the CAG repeat such as environmental and/or genetic factors.[39]
Background
Myotonic dystrophy is a rare muscular disorder in which numerous bodily systems are affected. There are four forms of Myotonic Dystrophy: mild phenotype and late-onset, onset in adolescence/young adulthood, early childhood featuring only learning disabilities, and a congenital form.[41] Individuals with Myotonic Dystrophy experience severe, debilitating physical symptoms such as muscle weakness, heartbeat issues, and difficulty breathing that can be improved through treatment to maximize patients' mobility and everyday activity to alleviate some stress of their caretakers.[42] The muscles of individuals with Myotonic Dystrophy feature an increase of type 1 fibers as well as an increased deterioration of these type 1 fibers.[42] In addition to these physical ailments, individuals with Myotonic Dystrophy have been found to experience varying internalized disorders such as anxiety and mood disorders as well as cognitive delays, attention deficit disorders, autism spectrum disorders, lower IQ's, and visual-spatial difficulties.[42] Research has shown that there is a direct correlation between expansion repeat number, IQ, and an individual's degree of visual-spatial impairment.[42]
Causation
Myotonic dystrophy results from a (CTG)n trinucleotide repeat expansion that resides in a 3' untranslated region of a serine/threonine kinase coding transcript.[43] This (CTG)n trinucleotide repeat is located within leukocytes; the length of the repeat and the age of the individual have been found to be directly related to disease progression and type 1 muscle fiber predominance.[43] Age and (CTG)n length only have small correlation coefficients to disease progression, research suggests that various other factors play a role in disease progression such as changes in signal transduction pathway, somatic expression, and cell heterogeneity in (CTG)n repeats.[43]
Background
Friedreich's ataxia is a progressive neurological disorder. Individuals experience gait and speech disturbances due to degeneration of the spinal cord and peripheral nerves. Other symptoms may include cardiac complications and diabetes. Typical age at symptom onset is 5–15, with symptoms progressively getting worse over time.[44]
Causation
Friedreich's ataxia is an autosomal recessive disorder cause by a GAA expansion in the intron of the FXN gene. This gene codes for the protein frataxin, a mitochondrial protein involved in iron homeostasis. The mutation impairs transcription of the protein, so affected cells produce only 5-10% of the frataxin of healthy cells.[45] This leads to iron accumulation in the mitochondria, and makes cells vulnerable to oxidative damage. Research shows that GAA repeat length is correlated with disease severity.[46]
Point of occurrence
Fragile X syndrome
The precise timing of TNR occurrence varies by disease. Although the exact timing for FXS is not certain, research has suggested that the earliest CGG expansions for this disorder are seen in primary oocytes.[47][48] It has been proposed that the repeat expansion happens in the maternal oocyte during meiotic cell cycle arrest in prophase I, however the mechanism remains nebulous.[49][50] Maternally inherited premutation alleles may expand into full mutation alleles (greater than 200 repeats), resulting in decreased production of the FMR-1 gene product FMRP and causing fragile X mental retardation syndrome.[51] For females, the large repeat expansions are based upon repair, while for males, the shortening of long repeat expansions is due to replication; therefore, their sperm lack these repeats, and paternal inheritance of long repeat expansions does not occur.[52][53][54] Between weeks 13 and 17 of human fetal development, the large CGG repeats are shortened.[54]
Myotonic dystrophy type 1
Many similarities can be drawn between DM1 and FXS involving aspects of mutation. Full maternal inheritance is present within DM1, repeat expansion length is linked to maternal age and the earliest instance of expansions is seen in the two-cell stage of preimplantation embryos.[55][56] There is a positive correlation between male inheritance and allele length.[57] A study of mice found the exact timing of CTG repeat expansion to be during development of spermatogonia.[58] In DM1 and FXS, it is hypothesized that expansion of TNRs occurs by means of multiple missteps by DNA polymerase in replication.[59][60] An inability of DNA polymerase to properly move across the TNR may cause transactivation of translesion polymerases (TLPs), which will attempt to complete the replication process and overcome the block. It is understood that as the DNA polymerase fails in this way, the resulting single-stranded loops left behind in the template strand undergo deletion, affecting TNR length. This process leaves the potential for TNR expansions to occur.[15]
Huntington's disease
In Huntington's disease (HD), the exact timing has not been determined; however there are a number of proposed points during germ cell development at which expansion is thought to occur.[61][62]
- In four HD samples examined, CAG repeat expansion lengths were more variable in mature sperm than that of sperm in development in the testes, leading to the conclusion that repeat expansions had a likelihood of occurring later in sperm development.[61]
- Repeat expansions have been observed to occur before the completion of meiosis in humans, specifically the first division.[63]
- In germ cells undergoing differentiation, evidence suggests it is possible for expansions to generate after the completion of meiosis as well, as larger HD mutations have been found in postmeiotic cells.[63][62]
Spinocerebellar ataxia type 1
Spinocerebellar ataxia type 1 (SCA1) CAG repeats are most often passed down through paternal inheritance and similarities can be seen with HD.[15] The tract size for offspring of mothers with these repeats does not display any degree of change.[64] Because TNR instability is not present in young female mice, and female SCA1 patient age and instability are directly related, expansions must occur in inactive oocytes.[65] A trend has seemed to emerge of larger expansions occurring in cells inactive in division and smaller expansions occurring in actively dividing or nondividing cells.[15]
Therapeutics
Trinucleotide repeat expansion, is a DNA mutation that is responsible for causing any type of disorder classified as a trinucleotide repeat disorder. These disorders are progressive and affect the sequences of the human genome, frequently within the nervous system. So far the available therapeutics only have modest results at best[66] with emphasis on the research and studying of genomic manipulation. The most advanced available therapies aim to target mutated gene expression by using antisense oligonucleotides (ASO) or RNA interference (RNAi) to target the messenger RNA (mRNA).[66] While solutions for the interventions of this disease is a priority, RNAi and ASO have only reached clinical trial stages.
RNA interference (RNAi)
RNA interference is a mechanism that can be used to silence the expression of genes, RNAi is a naturally occurring process that is leveraged using synthetic small interfering RNAs (siRNAs) that are used to change the action and duration of the natural RNAi process.[67] Another synthetic RNA is the short hairpin RNAs (shRNA)[67] these can also be used to monitor the action and predictability of the RNAi process.
RNAi begins with RNase Dicer cleaving a 21-25 nucleotide long stand of double stranded RNA substrates into small fragments. This process results in the creation of the siRNA duplexes that will be used by the complex RNA induced silencing complex (RISC).[67] The RISC contains the antisense that will bind to complementary mRNA strands, once they are bound they are cleaved by the protein found within the RISC complex called Argonaute 2 (Ago2) between the bases 10 and 11 relative to the 5' end. Before the cleavage of the mRNA strand the double stranded antisense of the siRNA is also cleaved by the Ago2 complex, this leaves a single stranded guide within the RISC compound that will be used to find the desired mRNA strand resulting in this process to have specificity.[68] Some problems that may occur is if the guide single strand siRNA within the RISC complex may become unstable when cleaved and begin to unwind, resulting in binding to an unfavorable mRNA strand. The perfect complementary guides for the targeted RNAs are easily recognized and will be cleaved within the RISC complex; if there is only partial complementary pairing between the guide strand and the targeted mRNA may cause the incorrect translation or destabilization at the target sites.[68]
Antisense oligonucleotides
Antisense oligonucleotides (ASOs) are small strand single stranded oligodeoxynucleotides approximately 15-20 nucleic acids in length that can alter the expression of a protein.[69] The goal of using these antisense oligonucleotides are the decrease in protein expression of a specific target usually by the inhibition of the RNase H endonuclease, as well as inhibition of the 5' cap formation or alteration of the splicing process.[70] In the native state ASOs are rapidly digested, this requires the use of phosphorylation order for the ASO to go through the cell membranes.
Despite the obvious benefits that antisense therapeutics can bring to the world with their ability to silence neural disease, there are many issues with the development of this therapy. One problem is the ASOs are highly susceptible to degradation by the nucleases[71] within the body. This results in a high amount of chemical modification when altering the chemistry to allow for the nucleases to surpass the degradation of these synthetic nucleic acids. Native ASOs have a very short half-life even before being filtered throughout the body especially in the kidney and with the a high negative charge makes the crossing through the vascular system or membranes very difficult when trying to reach the targeted DNA or mRNA strands. With all these barriers, the chemical modifications may lead to devastating effects when being introduced into the body making each problem develop more and more side effects.
The synthetic oligonucleotides are negatively charged molecules that are chemically modified in order for the molecule to regulate the gene expression within the cell. Some issues that come about this process is the toxicity and variability that can come about with chemical modification.[70] The goal of the ASO is to modulate the gene expression through proteins which can be done in 2 complex ways; a)the RNase H-dependent oligonucleotides, which induce the degradation of mRNA, and (b) the steric-blocker oligonucleotides, which physically prevent or inhibit the progression of splicing or the translational machinery. The majority of investigated ASOs utilize the first mechanism with the Rnase H enzyme that hydrolyzes an RNA strand, when this enzyme is assisted using the oligonucleotides the reduction of RNA expression is efficiently reduced by 80-95% and can still inhibit expression on any region of the mRNA.
References
- Richards RI, Sutherland GR (1997). "Dynamic mutation: possible mechanisms and significance in human disease". Trends Biochem. Sci. 22 (11): 432–6. doi:10.1016/S0968-0004(97)01108-0. PMID 9397685.
- Salinas-Rios V, Belotserkovskii BP, Hanawalt PC (2011). "DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability". Nucleic Acids Res. 39 (15): 1–11. doi:10.1093/nar/gkr429. PMC 3177194. PMID 21666257.
- McIvor EI, Polak U, Napierala M (2010). "New insights into repeat instability: Role of RNA•DNA hybrids". RNA Biol. 7 (5): 551–8. doi:10.4161/rna.7.5.12745. PMC 3073251. PMID 20729633.
- Usdin K, House NC, Freudenreich CH (2015). "Repeat instability during DNA repair: Insights from model systems". Crit. Rev. Biochem. Mol. Biol. 50 (2): 142–67. doi:10.3109/10409238.2014.999192. PMC 4454471. PMID 25608779.
- News Release, Weizmann Institute of Science, "Scientists at the Weizmann Institute, using computer simulations, have provided an explanation as to why certain genetic diseases caused by repeats in the code are "genetic time-bombs" whose onset and progression can be accurately predicted," November 21, 2007, at http://80.70.129.162/site/en/weizman.asp?pi=371&doc_id=5042 Archived 2011-05-31 at the Wayback Machine. Retrieved on 2007-12-30.
- Budworth, Helen; McMurray, Cynthia T. (2013). "A Brief History of Triplet Repeat Diseases". Trinucleotide Repeat Protocols. Methods in Molecular Biology. Vol. 1010. pp. 3–17. doi:10.1007/978-1-62703-411-1_1. ISBN 978-1-62703-410-4. ISSN 1064-3745. PMC 3913379. PMID 23754215.
- Verkerk, A. J.; Pieretti, M.; Sutcliffe, J. S.; Fu, Y. H.; Kuhl, D. P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M. F.; Zhang, F. P. (1991-05-31). "Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome". Cell. 65 (5): 905–914. doi:10.1016/0092-8674(91)90397-h. ISSN 0092-8674. PMID 1710175. S2CID 21463845.
- La Spada, A. R.; Wilson, E. M.; Lubahn, D. B.; Harding, A. E.; Fischbeck, K. H. (1991-07-04). "Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy". Nature. 352 (6330): 77–79. Bibcode:1991Natur.352...77S. doi:10.1038/352077a0. ISSN 0028-0836. PMID 2062380. S2CID 1678351.
- La Spada, Albert R.; Taylor, J. Paul (April 2010). "Repeat expansion disease: Progress and puzzles in disease pathogenesis". Nature Reviews. Genetics. 11 (4): 247–258. doi:10.1038/nrg2748. ISSN 1471-0056. PMC 4704680. PMID 20177426.
- "A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group". Cell. 72 (6): 971–983. 1993-03-26. doi:10.1016/0092-8674(93)90585-e. hdl:2027.42/30901. ISSN 0092-8674. PMID 8458085. S2CID 802885.
- Lee, Do-Yup; McMurray, Cynthia T. (June 2014). "Trinucleotide expansion in disease: why is there a length threshold?". Current Opinion in Genetics & Development. 26: 131–140. doi:10.1016/j.gde.2014.07.003. ISSN 0959-437X. PMC 4252851. PMID 25282113.
- Filla, A.; De Michele, G.; Cavalcanti, F.; Pianese, L.; Monticelli, A.; Campanella, G.; Cocozza, S. (September 1996). "The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia". American Journal of Human Genetics. 59 (3): 554–560. ISSN 0002-9297. PMC 1914893. PMID 8751856.
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M. (August 1993). "Trinucleotide repeat length instability and age of onset in Huntington's disease". Nature Genetics. 4 (4): 387–392. doi:10.1038/ng0893-387. ISSN 1061-4036. PMID 8401587. S2CID 7044715.
- Li, Jian-Liang; Hayden, Michael R; Warby, Simon C; Durr, Alexandra; Morrison, Patrick J; Nance, Martha; Ross, Christopher A; Margolis, Russell L; Rosenblatt, Adam; Squitieri, Ferdinando; Frati, Luigi (2006-08-17). "Genome-wide significance for a modifier of age at neurological onset in Huntington's Disease at 6q23-24: the HD MAPS study". BMC Medical Genetics. 7: 71. doi:10.1186/1471-2350-7-71. ISSN 1471-2350. PMC 1586197. PMID 16914060.
- McMurray, Cynthia T. (November 2010). "Mechanisms of trinucleotide repeat instability during human development". Nature Reviews. Genetics. 11 (11): 786–799. doi:10.1038/nrg2828. ISSN 1471-0064. PMC 3175376. PMID 20953213.
- Veitch, Nicola J.; Ennis, Margaret; McAbney, John P.; US-Venezuela Collaborative Research Project; Shelbourne, Peggy F.; Monckton, Darren G. (2007-06-01). "Inherited CAG.CTG allele length is a major modifier of somatic mutation length variability in Huntington disease". DNA Repair. 6 (6): 789–796. doi:10.1016/j.dnarep.2007.01.002. ISSN 1568-7864. PMID 17293170.
- Lee, J.-M.; Ramos, E.M.; Lee, J.-H.; Gillis, T.; Mysore, J.S.; Hayden, M.R.; Warby, S.C.; Morrison, P.; Nance, M.; Ross, C.A.; Margolis, R.L. (2012-03-06). "CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion". Neurology. 78 (10): 690–695. doi:10.1212/WNL.0b013e318249f683. ISSN 0028-3878. PMC 3306163. PMID 22323755.
- Bovo, D.; Rugge, M.; Shiao, Y. H. (February 1999). "Origin of spurious multiple bands in the amplification of microsatellite sequences". Molecular Pathology. 52 (1): 50–51. doi:10.1136/mp.52.1.50. ISSN 1366-8714. PMC 395672. PMID 10439841.
- Leeflang, E. P.; Zhang, L.; Tavaré, S.; Hubert, R.; Srinidhi, J.; MacDonald, M. E.; Myers, R. H.; de Young, M.; Wexler, N. S.; Gusella, J. F. (September 1995). "Single sperm analysis of the trinucleotide repeats in the Huntington's disease gene: quantification of the mutation frequency spectrum". Human Molecular Genetics. 4 (9): 1519–1526. doi:10.1093/hmg/4.9.1519. ISSN 0964-6906. PMID 8541834.
- Monckton, D. G.; Wong, L. J.; Ashizawa, T.; Caskey, C. T. (January 1995). "Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses". Human Molecular Genetics. 4 (1): 1–8. doi:10.1093/hmg/4.1.1. ISSN 0964-6906. PMID 7711720.
- Frey, Ulrich H.; Bachmann, Hagen S.; Peters, Jürgen; Siffert, Winfried (24 July 2008). "PCR-amplification of GC-rich regions: 'slowdown PCR'". Nature Protocols. 3 (8): 1312–1317. doi:10.1038/nprot.2008.112. ISSN 1750-2799. PMID 18714299. S2CID 22707736 – via Nature.
- Yoshinori, Kohwi (2013). Trinucleotide Repeat Protocols. New York: Humana Press. ISBN 978-1-62703-410-4.
- Ohshima, Keiichi; Wells, Robert D. (1997-07-04). "Hairpin Formation during DNA Synthesis Primer Realignmentin Vitro in Triplet Repeat Sequences from Human Hereditary Disease Genes". Journal of Biological Chemistry. 272 (27): 16798–16806. doi:10.1074/jbc.272.27.16798. ISSN 0021-9258. PMID 9201985. S2CID 22220445.
- Almeida, Bruno; Fernandes, Sara; Abreu, Isabel A.; Macedo-Ribeiro, Sandra (2013). "Trinucleotide Repeats: A Structural Perspective". Frontiers in Neurology. 4: 76. doi:10.3389/fneur.2013.00076. ISSN 1664-2295. PMC 3687200. PMID 23801983.
- Mirkin, Sergei M. (June 2007). "Expandable DNA repeats and human disease". Nature. 447 (7147): 932–940. Bibcode:2007Natur.447..932M. doi:10.1038/nature05977. ISSN 1476-4687. PMID 17581576. S2CID 4397592.
- Mosbach, Valentine; Poggi, Lucie; Richard, Guy-Franck (February 2019). "Trinucleotide repeat instability during double-strand break repair: from mechanisms to gene therapy". Current Genetics. 65 (1): 17–28. doi:10.1007/s00294-018-0865-1. ISSN 0172-8083. PMID 29974202. S2CID 49571068.
- Neil, Alexander J.; Kim, Jane C.; Mirkin, Sergei M. (September 2017). "Precarious maintenance of simple DNA repeats in eukaryotes". BioEssays. 39 (9): 1700077. doi:10.1002/bies.201700077. PMC 5577815. PMID 28703879.
- Xu, Pengning; Pan, Feng; Roland, Christopher; Sagui, Celeste; Weninger, Keith (2020-03-18). "Dynamics of strand slippage in DNA hairpins formed by CAG repeats: roles of sequence parity and trinucleotide interrupts". Nucleic Acids Research. 48 (5): 2232–2245. doi:10.1093/nar/gkaa036. ISSN 0305-1048. PMC 7049705. PMID 31974547.
- Mariappan, S V; Garcoa, A E; Gupta, G (1996-02-15). "Structure and dynamics of the DNA hairpins formed by tandemly repeated CTG triplets associated with myotonic dystrophy". Nucleic Acids Research. 24 (4): 775–783. doi:10.1093/nar/24.4.775. ISSN 0305-1048. PMC 145682. PMID 8604323.
- "Expansion Repeat Disorders". Expansion Therapeutics. Retrieved 2020-12-09.
- Liu, Vivian F.; Bhaumik, Dipa; Wang, Teresa S.-F. (February 1999). "Mutator Phenotype Induced by Aberrant Replication". Molecular and Cellular Biology. 19 (2): 1126–1135. doi:10.1128/mcb.19.2.1126. ISSN 0270-7306. PMC 116042. PMID 9891047.
- La Spada, A Trinucleotide Repeat Instability: Genetic Features and Molecular Mechanisms. Brain Pathology 1997; 7:943-963
- Fu Y.H. Kuhl, D.P. Piuuti, A. Pieretti, M. Sutcliffe, J.S. Richards, S. Verkerk, A.J. Holden, J.J. Fenwick, R.G. Jr. Warren, S.T. Oostra, B.A. Nelson, D.L. Caskey, C.T. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67:1047-58
- Ranen, N.G. Sine, O.C. Abbott, M.H. Sherr, M. Codori. A.M. Franz, M.L. Chao, N.I. Chung, A.S. Pleasant, N. Callahan, C. et al. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am J Hum Genet 1995; 57:593-602
- Tsiouris, J.A. Brown, W.T. Neuropsychiatric Symptoms of Fragile X Syndrome. CNS Drugs 2004; 18: 687–703
- Tassone, F. Hagerman, R.J. Loesch, D.Z. Lachiewics, A. Taylor, A.K. Hagerman, P.J. Fragile X Males With Unmethylated, Full Mutation Trinucleotide Repeat Expansions Have Elevated Levels of FMR1 Messenger RNA. American Journal of Medical Genetics 2000; 94: 232-236
- Nolin SL, Lewis French III A, Ye LL, et al. Familial transmission of the FMR1 CGG repeat. Am J Hum Genet 1996; 59: 1252–6
- Colak, D. Zaninovic, N. Cohen, M.S. Rosenwaks, Z. Yang, W. Gerhardt, J. Disney, M.D. Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014; 343(6174):1002-1005
- Panagopoulos, I. Lassen, C. Kristofferson, U. Åman, P. A Novel PCR-Based Approach for the Detection of the Huntington Disease Associated Trinucleotide Repeat Expansion. Human Mutation 1999; 13:232-236
- Laccone, F. Christian, W. A Recurrent Expansion of a Maternal Allele with 36 CAG Repeats Causes Huntington Disease in Two Sisters. American Journal of Human Genetics 2000. 66(3): 1145–1148
- Zeliha, U. Myotonic Dystrophy. Meandros Medical and Dental Journal 2019; 20(18):186-190
- Kledzik, A.M. Dunn, D.W. The importance of screening for internalizing symptoms, inattention, and cognitive difficulties in childhood‐onset myotonic dystrophy.
- Tohgi, H. Utsugisawa, K. Kawamorita, A. Yamagata, M. Saitoh, K. Hashimoto, K. Effects of CTG Trinucleotide Repeat Expansion in Leukocytes on Quantitative Muscle Histopathology in Myotonic Dystrophy. Muscle Nerve 1997; 20:232-234
- Cook, A.; Giunti, P. (2017). "Friedreich's ataxia: Clinical features, pathogenesis and management". British Medical Bulletin. 124 (1): 19–30. doi:10.1093/bmb/ldx034. PMC 5862303. PMID 29053830.
- Lazaropoulos M, Dong Y, Clark E, Greeley NR, Seyer LA, Brigatti KW, et al. (August 2015). "Frataxin levels in peripheral tissue in Friedreich ataxia". Annals of Clinical and Translational Neurology. 2 (8): 831–42. doi:10.1002/acn3.225. PMC 4554444. PMID 26339677.
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, et al. (October 1996). "Clinical and genetic abnormalities in patients with Friedreich's ataxia". The New England Journal of Medicine. 335 (16): 1169–75. doi:10.1056/nejm199610173351601. PMID 8815938.
- Rifé, M.; Badenas, C.; Quintó, Ll; Puigoriol, E.; Tazón, B.; Rodriguez-Revenga, L.; Jiménez, L.; Sánchez, A.; Milà, M. (October 2004). "Analysis of CGG variation through 642 meioses in Fragile X families". Molecular Human Reproduction. 10 (10): 773–776. doi:10.1093/molehr/gah102. ISSN 1360-9947. PMID 15322225.
- Sermon, K.; Seneca, S.; Vanderfaeillie, A.; Lissens, W.; Joris, H.; Vandervorst, M.; Van Steirteghem, A.; Liebaers, I. (December 1999). "Preimplantation diagnosis for fragile X syndrome based on the detection of the non-expanded paternal and maternal CGG". Prenatal Diagnosis. 19 (13): 1223–1230. doi:10.1002/(SICI)1097-0223(199912)19:13<1223::AID-PD724>3.0.CO;2-0. ISSN 0197-3851. PMID 10660959. S2CID 40581707.
- Tripathi, Anima; Kumar, K. V. Prem; Chaube, Shail K. (June 2010). "Meiotic cell cycle arrest in mammalian oocytes". Journal of Cellular Physiology. 223 (3): 592–600. doi:10.1002/jcp.22108. ISSN 1097-4652. PMID 20232297. S2CID 33819727.
- McMurray, Cynthia T. (November 2010). "Mechanisms of trinucleotide repeat instability during human development". Nature Reviews. Genetics. 11 (11): 786–799. doi:10.1038/nrg2828. ISSN 1471-0056. PMC 3175376. PMID 20953213.
- Entezam, Ali; Biacsi, Rea; Orrison, Bonnie; Saha, Tapas; Hoffman, Gloria E.; Grabczyk, Ed; Nussbaum, Robert L.; Usdin, Karen (2007-06-15). "Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model". Gene. 395 (1–2): 125–134. doi:10.1016/j.gene.2007.02.026. ISSN 0378-1119. PMC 1950257. PMID 17442505.
- Fu, Y. H.; Kuhl, D. P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J. S.; Richards, S.; Verkerk, A. J.; Holden, J. J.; Fenwick, R. G.; Warren, S. T. (1991-12-20). "Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox". Cell. 67 (6): 1047–1058. doi:10.1016/0092-8674(91)90283-5. ISSN 0092-8674. PMID 1760838. S2CID 21970859.
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A. J.; Jorens, H. Z.; Darby, J. K.; Oostra, B. (June 1993). "The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm". Nature Genetics. 4 (2): 143–146. doi:10.1038/ng0693-143. ISSN 1061-4036. PMID 8348152. S2CID 9857049.
- Malter, H. E.; Iber, J. C.; Willemsen, R.; de Graaff, E.; Tarleton, J. C.; Leisti, J.; Warren, S. T.; Oostra, B. A. (February 1997). "Characterization of the full fragile X syndrome mutation in fetal gametes". Nature Genetics. 15 (2): 165–169. doi:10.1038/ng0297-165. ISSN 1061-4036. PMID 9020841. S2CID 2836586.
- Dean, Nicola L.; Tan, Seang Lin; Ao, Asangla (July 2006). "Instability in the transmission of the myotonic dystrophy CTG repeat in human oocytes and preimplantation embryos". Fertility and Sterility. 86 (1): 98–105. doi:10.1016/j.fertnstert.2005.12.025. ISSN 1556-5653. PMID 16716318.
- Temmerman, Nele De; Sermon, Karen; Seneca, Sara; De Rycke, Martine; Hilven, Pierre; Lissens, Willy; Van Steirteghem, André; Liebaers, Inge (August 2004). "Intergenerational Instability of the Expanded CTG Repeat in the DMPK Gene: Studies in Human Gametes and Preimplantation Embryos". American Journal of Human Genetics. 75 (2): 325–329. doi:10.1086/422762. ISSN 0002-9297. PMC 1216067. PMID 15185171.
- Martorell, L.; Gámez, J.; Cayuela, M. L.; Gould, F. K.; McAbney, J. P.; Ashizawa, T.; Monckton, D. G.; Baiget, M. (2004-01-27). "Germline mutational dynamics in myotonic dystrophy type 1 males: allele length and age effects". Neurology. 62 (2): 269–274. doi:10.1212/wnl.62.2.269. ISSN 1526-632X. PMID 14745066. S2CID 28115656.
- Savouret, Cédric; Garcia-Cordier, Corinne; Megret, Jérôme; te Riele, Hein; Junien, Claudine; Gourdon, Geneviève (January 2004). "MSH2-Dependent Germinal CTG Repeat Expansions Are Produced Continuously in Spermatogonia from DM1 Transgenic Mice". Molecular and Cellular Biology. 24 (2): 629–637. doi:10.1128/MCB.24.2.629-637.2004. ISSN 0270-7306. PMC 343816. PMID 14701736.
- Maul, Robert W.; Sutton, Mark D. (November 2005). "Roles of the Escherichia coli RecA Protein and the Global SOS Response in Effecting DNA Polymerase Selection In Vivo". Journal of Bacteriology. 187 (22): 7607–7618. doi:10.1128/JB.187.22.7607-7618.2005. ISSN 0021-9193. PMC 1280315. PMID 16267285.
- Guo, Caixia; Kosarek-Stancel, J. Nicole; Tang, Tie-Shan; Friedberg, Errol C. (July 2009). "Y-family DNA polymerases in mammalian cells". Cellular and Molecular Life Sciences. 66 (14): 2363–2381. doi:10.1007/s00018-009-0024-4. ISSN 1420-9071. PMID 19367366. S2CID 8350634.
- Telenius, H.; Kremer, B.; Goldberg, Y. P.; Theilmann, J.; Andrew, S. E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E. J.; Clarke, L. A. (April 1994). "Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm". Nature Genetics. 6 (4): 409–414. doi:10.1038/ng0494-409. ISSN 1061-4036. PMID 8054984. S2CID 22191363.
- Leeflang, E. P.; Tavaré, S.; Marjoram, P.; Neal, C. O.; Srinidhi, J.; MacFarlane, H.; MacDonald, M. E.; Gusella, J. F.; de Young, M.; Wexler, N. S.; Arnheim, N. (February 1999). "Analysis of germline mutation spectra at the Huntington's disease locus supports a mitotic mutation mechanism". Human Molecular Genetics. 8 (2): 173–183. doi:10.1093/hmg/8.2.173. ISSN 0964-6906. PMID 9931325.
- Yoon, Song-Ro; Dubeau, Louis; de Young, Margot; Wexler, Nancy S.; Arnheim, Norman (July 22, 2003). "Huntington disease expansion mutations in humans can occur before meiosis is completed". Proceedings of the National Academy of Sciences of the United States of America. 100 (15): 8834–8838. Bibcode:2003PNAS..100.8834Y. doi:10.1073/pnas.1331390100. ISSN 0027-8424. PMC 166399. PMID 12857955.
- Koefoed, P.; Hasholt, L.; Fenger, K.; Nielsen, J. E.; Eiberg, H.; Buschard, K.; Sørensen, S. A. (November 1998). "Mitotic and meiotic instability of the CAG trinucleotide repeat in spinocerebellar ataxia type 1". Human Genetics. 103 (5): 564–569. doi:10.1007/s004390050870. ISSN 0340-6717. PMID 9860298. S2CID 20659803.
- Kaytor, M. D.; Burright, E. N.; Duvick, L. A.; Zoghbi, H. Y.; Orr, H. T. (November 1997). "Increased trinucleotide repeat instability with advanced maternal age". Human Molecular Genetics. 6 (12): 2135–2139. doi:10.1093/hmg/6.12.2135. ISSN 0964-6906. PMID 9328478.
- Gonzalez-Alegre, Pedro (2019-10-01). "Recent advances in molecular therapies for neurological disease: triplet repeat disorders". Human Molecular Genetics. 28 (R1): R80–R87. doi:10.1093/hmg/ddz138. ISSN 0964-6906. PMC 6796999. PMID 31227833.
- Bumcrot, David; Manoharan, Muthiah; Koteliansky, Victor; Sah, Dinah W. Y. (December 2006). "RNAi therapeutics: a potential new class of pharmaceutical drugs". Nature Chemical Biology. 2 (12): 711–719. doi:10.1038/nchembio839. ISSN 1552-4469. PMC 7097247. PMID 17108989.
- Aagaard, Lars; Rossi, John J. (2007-03-30). "RNAi Therapeutics: Principles, Prospects and Challenges". Advanced Drug Delivery Reviews. 59 (2–3): 75–86. doi:10.1016/j.addr.2007.03.005. ISSN 0169-409X. PMC 1978219. PMID 17449137.
- Rinaldi, Carlo; Wood, Matthew J. A. (January 2018). "Antisense oligonucleotides: the next frontier for treatment of neurological disorders". Nature Reviews Neurology. 14 (1): 9–21. doi:10.1038/nrneurol.2017.148. ISSN 1759-4766. PMID 29192260. S2CID 28579577.
- Di Fusco, Davide; Dinallo, Vincenzo; Marafini, Irene; Figliuzzi, Michele M.; Romano, Barbara; Monteleone, Giovanni (2019). "Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease". Frontiers in Pharmacology. 10: 305. doi:10.3389/fphar.2019.00305. ISSN 1663-9812. PMC 6450224. PMID 30983999.
- Verma, Ashok (2018). "Recent Advances in Antisense Oligonucleotide Therapy in Genetic Neuromuscular Diseases". Annals of Indian Academy of Neurology. 21 (1): 3–8. doi:10.4103/aian.AIAN_298_17. ISSN 0972-2327. PMC 5909143. PMID 29720791.