VLDL receptor
The very-low-density-lipoprotein receptor (VLDLR) is a transmembrane lipoprotein receptor of the low-density-lipoprotein (LDL) receptor family. VLDLR shows considerable homology with the members of this lineage. Discovered in 1992 by T. Yamamoto, VLDLR is widely distributed throughout the tissues of the body, including the heart, skeletal muscle, adipose tissue, and the brain, but is absent from the liver.[5] This receptor has an important role in cholesterol uptake, metabolism of apolipoprotein E-containing triacylglycerol-rich lipoproteins, and neuronal migration in the developing brain. In humans, VLDLR is encoded by the VLDLR gene. Mutations of this gene may lead to a variety of symptoms and diseases, which include type I lissencephaly, cerebellar hypoplasia, and atherosclerosis.
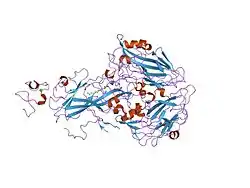
Protein structure
VLDLR is a member of the low-density-lipoprotein (LDL) receptor family, which is entirely composed of type I transmembrane lipoprotein receptors.
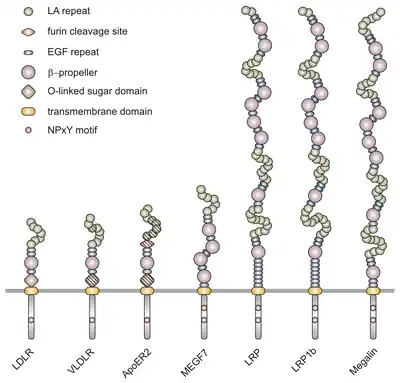
All members of this family share five highly conserved structural domains: an extracellular N-terminal ligand-binding domain with cysteine-rich repeats (also called ligand-binding repeats), an epidermal growth factor (EGF), an O-linked glycosylation sugar domain, a single transmembrane sequence, and a cytoplasmic domain which contains an NPxY sequence. The NPxY motif functions in signal transduction and the targeting of receptors to coated pits and consists of the sequence Asparagine-Proline-X-Tyrosine, where X can be any amino acid.[6] Mimicking this general structure, VLDLR has eight, 40 amino acid long cysteine-rich repeats in its extracellular N-terminal ligand-binding domain.[6] This is the main difference from the main member of the LDL receptor family, LDLR, which has only seven cysteine-rich repeats which are also 40 amino acids long.[7] Each of these cysteine-rich repeats, in both VLDLR and LDLR, has three disulfide bonds and a coordinated Ca2+ ion. The N-terminus also consists of a glycine residue followed by 27 hydrophobic residues that constitute the signal peptide.[6] Following this region is an EGF repeat, a β-propeller segment that plays a role in the pH-dependent dissociation of the ligand-receptor complex,[8] and two more EGF repeats.[9] The VLDLR O-linked glycosylation domain, next in the sequence, has many threonine and serine residues and totals 46 amino acids. The transmembrane domain, which functions in anchoring the receptors to the membrane, is 22 amino acids long.[6] Final in the sequence is the 54 amino acid cytoplasmic domain, which contains the NPxY motif.[8]
Isoforms
The full-length human VLDLR genome is located on locus 9p24 on chromosome 9. It consists of a 40 kb segment that includes 19 exon-coding sequences, which is one more exon than encoded by LDLR. This extra exon in the VLDLR gene accounts for the extra cysteine-binding repeat not found in LDLR.[7] Together, the exons making up the VLDLR gene encode a protein that is 873 amino acid residues long. VLDLR is known to exist as four different protein isoforms: type I, II, III, and IV. These different isoforms result from variations in alternative splicing. The transcript of type I VLDLR (VLDLR-I) is composed of all 19 exons. VLDLR-II, on the other hand, lacks exon 16, which encodes for the O-glycosylation domain between sugar regions. VLDLR-III lacks exon 4 that encodes the third ligand-binding repeat. Finally, VLDLR-IV transcripts lack both exon 16 and exon 4. It has been shown that 75% of VLDLR transcripts exist as isoform type II in mouse brain models. This shows that most VLDLRs in the brain are not glycosylated, as type II lacks exon 16 which encodes the O-glycosylation domain. Isoform type IV is known to be the second most prominent.[6]
Evolutionary conservation
There is a high level of conservation within the LDL receptor family. In particular, there is 50% overall sequence homology between VLDLR and ApoER2, another lipoprotein receptor of this family.[6] Comparing LDLR and VLDLR, it was found that their primary structures are 55% identical within their ligand-binding regions. The modular structures of these two proteins are almost superimposable, with the only difference being the additional cysteine-rich repeat in VLDLR. This is demonstrated through the alignment of the two receptors according to their linker region; in LDLR, the linker region is located between cysteine-rich repeats four and five of its seven repeats while in VLDLR, the linker region appears to be between repeats five and six of its eight repeats.[10]
VLDLR also shows high homology among various species. VLDLR of humans, mice, rats, and rabbits have been identified as 95% identical. Furthermore, there is approximately 84% conservation with the respective protein in chickens. This level of homology between species is much higher than that found for LDLR. Hence, these gene comparisons suggest that VLDLR and LDLR diverged before the LDLRs did among vertebrates.[10]
Ligand binding
VLDLR binds compounds containing apolipoprotein E (apoE). These ligands attach to the cysteine binding repeats in the N-terminus end. The difference in cysteine-rich repeats between the members of the LDL receptor family lead to the differences in binding affinity. VLDLR, in particular, binds VLDL and intermediate-density lipoprotein (IDL), but not LDL. This inability to bind LDL is due to VLDLR's incapability to bind apolipoprotein B (apoB), which is present in LDL.[11]
Inhibitors
Receptor-associated protein (RAP) and thrombospondin-1 (THBS1) have been identified as compounds that bind VLDLR. In many cases, these compounds exhibit inhibitory effects. THBS1 binds VLDLR and blocks ligand binding.[11] This plays an important role in the reelin pathway, as THBS1 can block the attachment of reelin, while simultaneously stimulating the transcription factors normally activated by reelin. This binding of THBS1, however, does not induce the subsequent degradation of these transcription factors, as reelin does, and can thus lead to greatly amplified effects.[6] The RAP protein acts similarly by blocking reelin from binding VLDLR. However, in this case phosphorylation of transcription factors, usually performed by reelin, is also blocked.[12]
Tissue distribution and expression
VLDLR is found throughout the body, with particularly high expression in fatty acid tissues due to their high level of triglycerides, VLDLR’s primary ligand. These tissues include those of the heart, skeletal muscle, and adipose layer. In addition, the receptor is found in macrophages, endothelial cells of capillaries,[8] and in the brain, where it has a very different function from that found in the rest of the body. There is a preferred expression for VLDLR type I in the heart, skeletal muscle and brain, as opposed to type II, which is mainly expressed in non-muscular tissues including the cerebrum, cerebellum, kidney, spleen, and aortic endothelial cells.[7][11] The highest expression of VLDLR is found in the brain. Although VLDLR is found in almost all regions of the brain, its highest expression is restricted to the cortex and cerebellum. Here, the receptor can be found on resting or activated microglia that are associated with senile plaques and cortical neurons, neuroblasts, matrix cells, Cajal-Retzius cells, glioblasts, astrocytes, oligodendrocytes, and region-specific pyramidal neurons.[6] Despite its major role in cholesterol and fatty acid metabolism, VLDLR is not found in the liver. This phenomenon is mainly attributed to the very high levels of LDLR in these areas.[7] In addition, it has been uncovered that this receptor is found, sub-cellularly, in the non-lipid raft sections of cell membranes.[6]
Regulation
Unlike LDLR, VLDLR does not exhibit any feedback mechanism, and hence intracellular lipoproteins are incapable of regulating it. This phenomenon is due to a difference in the sterol regulatory element-1 (SRE-1) of VLDLR. Normal SRE-1 sequences, like those found in LDLR, are characterized by two repeats of the codon CAC separated by two intervening C nucleotides (5’-CACCCCAC-3’). The sterol regulatory element-binding protein-1 (SREBP-1), a transcription factor, targets the CAC repeats of SRE-1 to regulate the protein’s transcription. However, the VLDLR gene is encoded by two SRE-1-like sequences that contain single nucleotide polymorphisms. These polymorphisms disrupt the SREBP-1 binding to the CAC repeats, and hence eliminate the feedback mechanism seen in other proteins.[7]
VLDLR expression is regulated by peroxisome proliferator-activated receptor-gamma (PPAR-γ). A 2010 study showed that the prescription drug Pioglitazone, an agonist of PPAR-γ, increases VLDLR mRNA expression and protein levels in experiments using mouse fibroblasts. The Pioglitazone treated mice exhibited a higher conversion rate of plasma triglycerides into epididymal fats. As expected, mice deficient in VLDLR did not show this same response.[8] These results suggest that VLDLR is important in fat accumulation.[8]
Many other hormones and dietary factors also regulate VLDLR expression. Thyroid hormone positively regulates VLDLR expression in skeletal muscles of rats, but not in adipose or heart tissues. In rabbits, VLDLR expression in heart muscle is up-regulated by estrogen and down-regulated by granulocyte-macrophage colony-stimulating factor. In trophoblast-derived cell lines, up-regulated VLDLR expression occurs when cells are incubated with hypolipidemic agents such as insulin and clofibrate. In contrast, 8-bromoadenosine 3',5'-cyclic monophosphate (8-bromo-cAMP) down-regulates VLDLR expression. Finally, VLDLR is affected by the presence of apoE and LDLR. The presence of apoE is required for VLDLR expression regulation, while the absence of LDLR alters the sterol-regulatory-element-1-like sequences of VLDLR to make them functional in only heart and skeletal muscle.[7]
Function
Beyond the nervous system
VLDLR is a peripheral lipoprotein receptor that functions in lipoprotein metabolism, cardiac fatty acid metabolism, and fat deposition. In effect, VLDLR will allow cholesterol to reach tissues from the bloodstream, where it may be used in cellular membranes. In addition, it will allow fatty acids to get into cells where they may be used as an energy source.[7] Overall, VLDLR primarily modulates the extra-hepatic metabolism of triglyceride-rich lipoproteins.[8]
Lipoprotein uptake
VLDLR only plays a discrete role in lipid metabolism, but is more significant in stressed situations. Mice with double knockouts in VLDLR and LDLR have higher serum triglyceride levels than those with only a knockout in the LDLR gene. In addition, LDLR knockout mice overexpressing VLDLR have decreased serum triglyceride levels. Although fat deposition is close to normal without VLDLR, its role gains importance when LDLR is deficient. Despite this knowledge on its role in lipoprotein uptake, the complete mechanism of lipid metabolism performed by VLDLR is not fully understood.[11]
Endocytosis
VLDLR is known to employ endocytosis, although the exact mechanism of this process is unknown for this protein. Endocytosis is mediated through NPxY sequences known to signal for receptor internalization through clathrin-coated pits. The presence of this sequence in the cytoplasmic tail of VLDLR makes endocytosis possible.[11] In general, lipoprotein receptors undergo a process by which they are endocytosed with their ligand into clathrin-coated pits. From here, they are together transported to early and late endosomes until reaching the lysosome. At this point, hydrolysis occurs and lipoprotein is released into the cytoplasm while the receptors are recycled back to the cell surface. It is not yet confirmed if VLDLR follows this exact mechanism, but one closely related to it is likely.[8]
In the nervous system
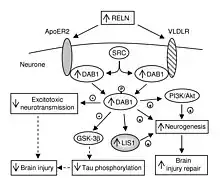
In addition to its role throughout the body, VLDLR has a unique role in the brain. It is a key component of the reelin pathway, which functions on one hand side in neuronal migration during the development of the brain, on the other hand in the retention of new memory traces in the hippocampal formation.[13][14] VLDLR links the reelin protein to an intracellular signaling protein, Dab1, that tells the individual neurons where to go within the anatomy of the brain. Mutations in VLDLR often do not lead to major disorganization as seen in reelin mutations. However, a VLDLR mutation does lead to some disorganization primarily located in the cerebellum, where VLDLR is believed to be most prominent.[6]
Neuronal migration
VLDLR is expressed on migrating neurons to help guide them to their proper location in the brain. This process is part of the reelin pathway, which is responsible for the inside-out formation of the six-layered neocortex.[6] Despite the discovery of this pathway, many of the specifics and molecular mechanisms of this process are still being debated. The presence of two reelin receptors, VLDLR and ApoER2, has made it difficult to distinguish each protein’s specific function.[15]
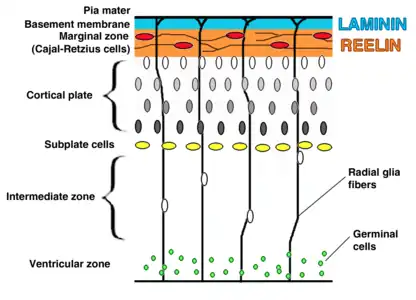
VLDLR is primarily responsible for the correct layering of pyramidal cells into layer 1 of the cerebral cortex. In particular, the absence of VLDLR may lead to ectopic accumulation of pyramidal cells in this region.[15] VLDLR does not affect the migration of early born cells into an organized layer, but since its absence results in the invasion of these neuroblasts into the marginal zone, it is theorized that VLDLR may encode a “stop signal.” This is supported by the fact that VLDLR is primarily expressed in the cortical plate adjacent to reelin-expressing cells, Cajal–Retzius cells, and in the intermediate zone. However, definitive evidence has not yet been found.[6] In general, reelin binds VLDLR and undergoes endocytosis via clathrin-coated vesicles.[6] Meanwhile, an intracellular protein, Dab1, has a PI/PTB domain that interacts with the NPxY sequence found in the cytoplasmic tail of VLDLR.[12] As a result, Dab1 is tyrosine phosphorylated and reelin is degraded. Finally, phosphorylated Dab1 activates an intracellular signaling cascade that directs neuroblasts to their proper location through the alteration of the cytoskeleton.[12][16] Many of the specifics of this pathway are still being investigated. It is not yet known if Dab1 is phosphorylated as a result of the endocytosis of reelin, or if there is another mechanism at play. In addition to the organization of the neocortex, VLDLR also plays a role in neuronal migration of the hippocampus and the Purkinje cells of the cerebellum. Yet, much information on this process is still unknown.[6]
Associated disorders
Mutations within the VLDLR gene lead to a multitude of disorders of varying severities. These disorders are usually associated with cholesterol homeostasis or a disorganization of neuron ordering in the brain due to disruption of the reelin pathway. The most prominent of these diseases are type I lissencephaly, VLDR-associated cerebellar hypoplasia, and atherosclerosis. In contrast to causing diseases, VLDLR has also been identified as a possible remedy for some disorders. Implementation of VLDLR into the liver may cure familial hypercholesterolemia (FH) in patients who either have defective LDLR or have defective immune systems that attack this protein. Since VLDLR is non-immunogenic it does not initiate an immune response, thus it is able to function normally under defective immune systems.[7] In addition, being that apoE, a major ligand of VLDLR, is a leading genetic risk factor for Alzheimer’s disease, VLDLR may play a role in modulating the risk of this disorder [6] which is explained by the fact that a decrease in reelin signaling in the fascia dentata is supposed to initiate Alzheimer's disease.[17] VLDLR has also been shown to reduce the chances of premature heart disease and stroke because VLDLR clears out lipoprotein A (Lp(a)), a major inherited risk factor for these diseases.[7]
Type 1 lissencephaly
Type I lissencephaly, or agyria-pachygyria, is a rare developmental disorder characterized by the absence of gyri and sulci in the brain. These severe malformations are a result of aberrant neuronal migration. In classical type I lissencephaly, neuronal migration begins but is unable to continue to completion. This process is likely disrupted by alterations to several genes, including the VLDLR, DCX, ARX, TUBA1A, RELN and LIS1. The severity of type I lissencephaly therefore varies with the mutation type. A homozygous deletion affecting the VLDLR gene results in a low degree of cortical thickening and absence of a cell-sparse zone. The cell-sparse zone describes the region between the outer and inner cortical layers of arrested neurons.[18] In addition, type 1 lissencephaly is closely associated with cerebellar hypoplasia.
VLDLR-associated cerebellar hypoplasia
Disequilibrium syndrome (DES) was first described in the 1970s as a non-progressive, neurological disorder.[19] In a 2005 study, DES was renamed as VLDLR-associated cerebellar hypoplasia (VLDLRCH) after its cause was linked to a disruption in the VLDLR gene.[20] At least six mutations affecting the homozygous recessive allele of the VLDLR gene have been identified and found to cause VLDLRCH. Several of these mutations have been localized to specific exons encoding the gene. One such mutation is a cytosine to thymine transition at base pair 1342 in exon 10 that causes a substitution at Arg448 for a termination signal. Likewise, there is evidence of a cytosine to thymine transition at base pair number 769 in exon 5 that causes a substitution at Arg257 for a termination signal. A third known mutation is caused by a homozygous 1-base pair deletion in exon 17 that causes a frameshift and premature termination in the O-linked sugar domain.[21] All such alterations to the VLDLR gene prevent the production of VLDLR and are therefore termed loss-of-function mutations. The recognized symptoms of VLDLRCH are moderate-to-severe intellectual disability, seizures, dysarthria, strabismus and delayed locomotion. In some cases, children with VLDLRCH learn to walk very late in development after the age of six years, or never learn to walk independently. The frequency of this disorder is unknown because early diagnosis of VLDLRCH is difficult using imaging techniques. It is associated with parental consanguinity and found in secluded communities such as the Hutterites and inbred families from Iran and Turkey.[22]
Atherosclerosis
Atherosclerosis is marked by an excessive accumulation of cholesterol by macrophages, leading to their transformation into foam cells. This accumulation of cholesterol is caused by dysregulation of cholesterol influx and efflux. Since macrophages do not have the ability to limit the influx of cholesterol, the balance is completely dependent on efflux pathways. VLDLR is expressed by macrophages, and functions in the uptake of native lipoproteins. Uniquely, VLDLR does not respond to cholesterol loading, likely due to its lack of feedback mechanisms. The inability to control its uptake of native lipoproteins makes VLDLR a pro-atherogenic factor.[23] This characteristic is supported by results from a 2005 study, in which reintroduction of VLDLR into VLDLR knockout mice led to greatly increased atherosclerotic lesion development.[23]
See also
References
- GRCh38: Ensembl release 89: ENSG00000147852 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000024924 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Nimpf J, Schneider WJ (December 2000). "From cholesterol transport to signal transduction: low density lipoprotein receptor, very low density lipoprotein receptor, and apolipoprotein E receptor-2". Biochim. Biophys. Acta. 1529 (1–3): 287–98. doi:10.1016/S1388-1981(00)00155-4. PMID 11111096.
- Reddy SS, Connor TE, Weeber EJ, Rebeck W (2011). "Similarities and differences in structure, expression, and functions of VLDLR and ApoER2". Mol Neurodegener. 6: 30. doi:10.1186/1750-1326-6-30. PMC 3113299. PMID 21554715.
- Takahashi S, Sakai J, Fujino T, Hattori H, Zenimaru Y, Suzuki J, Miyamori I, Yamamoto TT (2004). "The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor". J. Atheroscler. Thromb. 11 (4): 200–8. doi:10.5551/jat.11.200. PMID 15356379.
- Go GW, Mani A (March 2012). "Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis". Yale J Biol Med. 85 (1): 19–28. PMC 3313535. PMID 22461740.
- Tissir F, Goffinet AM (June 2003). "Reelin and brain development". Nat. Rev. Neurosci. 4 (6): 496–505. doi:10.1038/nrn1113. PMID 12778121. S2CID 12039624.
- Nimpf J, Schneider WJ (December 1998). "The VLDL receptor: an LDL receptor relative with eight ligand binding repeats, LR8". Atherosclerosis. 141 (2): 191–202. doi:10.1016/s0021-9150(98)00172-5. PMID 9862168.
- GUPEA: Mechanisms for and consequences of cellular lipid accumulation - Role of the Very Low Density Lipoprotein (VLDL) receptor. 2011-12-02. hdl:2077/27815. ISBN 9789162883560.
- Rice DS, Curran T (2001). "Role of the reelin signaling pathway in central nervous system development". Annu. Rev. Neurosci. 24: 1005–39. doi:10.1146/annurev.neuro.24.1.1005. PMID 11520926. S2CID 17258257.
- Kovács KA (September 2020). "Episodic Memories: How do the Hippocampus and the Entorhinal Ring Attractors Cooperate to Create Them?". Frontiers in Systems Neuroscience. 14: 68. doi:10.3389/fnsys.2020.559186. PMC 7511719. PMID 33013334.
- Kovács KA (December 2021). "Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer's Disease". International Journal of Molecular Sciences. 23 (1): 462. doi:10.3390/ijms23010462. PMC 8745479. PMID 35008886.
- Valiente M, Marín O (February 2010). "Neuronal migration mechanisms in development and disease". Curr. Opin. Neurobiol. 20 (1): 68–78. doi:10.1016/j.conb.2009.12.003. PMID 20053546. S2CID 18658808.
- Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG (2004). "Cortical neuronal migration mutants suggest separate but intersecting pathways". Annu. Rev. Cell Dev. Biol. 20: 593–618. doi:10.1146/annurev.cellbio.20.082503.103047. PMID 15473853.
- Kovács KA (December 2021). "Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer's Disease". International Journal of Molecular Sciences. 23 (1): 462. doi:10.3390/ijms23010462. PMC 8745479. PMID 35008886.
- Spalice A, Parisi P, Nicita F, Pizzardi G, Del Balzo F, Iannetti P (March 2009). "Neuronal migration disorders: clinical, neuroradiologic and genetics aspects". Acta Paediatr. 98 (3): 421–33. doi:10.1111/j.1651-2227.2008.01160.x. PMID 19120042. S2CID 21620197.
- Moheb LA, Tzschach A, Garshasbi M, Kahrizi K, Darvish H, Heshmati Y, Kordi A, Najmabadi H, Ropers HH, Kuss AW (February 2008). "Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome". Eur. J. Hum. Genet. 16 (2): 270–3. doi:10.1038/sj.ejhg.5201967. PMID 18043714.
- Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS (September 2005). "Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification". Am. J. Hum. Genet. 77 (3): 477–83. doi:10.1086/444400. PMC 1226212. PMID 16080122.
- Online Mendelian Inheritance in Man (OMIM): Cerebellar Hypoplasia, VLDLR-Associated; VLDLRCH - 224050
- Boycott KM, Parboosingh JS (2008). "VLDLR-Associated Cerebellar Hypoplasia". In Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds.). GeneReviews [Internet]. University of Washington, Seattle. PMID 20301729.
- Pennings M, Meurs I, Ye D, Out R, Hoekstra M, Van Berkel TJ, Van Eck M (October 2006). "Regulation of cholesterol homeostasis in macrophages and consequences for atherosclerotic lesion development". FEBS Lett. 580 (23): 5588–96. doi:10.1016/j.febslet.2006.08.022. PMID 16935283. S2CID 42158329.
Further reading
- Oka K, Ishimura-Oka K, Chu MJ, Sullivan M, Krushkal J, Li WH, Chan L (September 1994). "Mouse very-low-density-lipoprotein receptor (VLDLR) cDNA cloning, tissue-specific expression and evolutionary relationship with the low-density-lipoprotein receptor". Eur. J. Biochem. 224 (3): 975–82. doi:10.1111/j.1432-1033.1994.00975.x. PMID 7925422.
- Ananyeva NM, Makogonenko YM, Kouiavskaia DV, Ruiz J, Limburg V, Meijer AB, Khrenov AV, Shima M, Strickland DK, Saenko EL (March 2008). "The binding sites for the very low density lipoprotein receptor and low-density lipoprotein receptor-related protein are shared within coagulation factor VIII". Blood Coagul. Fibrinolysis. 19 (2): 166–77. doi:10.1097/MBC.0b013e3282f5457b. PMID 18277139. S2CID 10380641.
- Ananyeva NM, Makogonenko YM, Sarafanov AG, Pechik IV, Gorlatova N, Radtke KP, Shima M, Saenko EL (September 2008). "Interaction of coagulation factor VIII with members of the low-density lipoprotein receptor family follows common mechanism and involves consensus residues within the A2 binding site 484-509". Blood Coagul. Fibrinolysis. 19 (6): 543–55. doi:10.1097/MBC.0b013e3283068859. PMID 18685438. S2CID 31127950.
- Llorca J, Rodríguez-Rodríguez E, Dierssen-Sotos T, Delgado-Rodríguez M, Berciano J, Combarros O (January 2008). "Meta-analysis of genetic variability in the beta-amyloid production, aggregation and degradation metabolic pathways and the risk of Alzheimer's disease". Acta Neurol. Scand. 117 (1): 070914011339003––. doi:10.1111/j.1600-0404.2007.00899.x. PMID 17854420. S2CID 25781860.
- Ozcelik T, Akarsu N, Uz E, Caglayan S, Gulsuner S, Onat OE, Tan M, Tan U (March 2008). "Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans". Proc. Natl. Acad. Sci. U.S.A. 105 (11): 4232–6. Bibcode:2008PNAS..105.4232O. doi:10.1073/pnas.0710010105. PMC 2393756. PMID 18326629.
- Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S (September 2008). "Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene". Eur. J. Hum. Genet. 16 (9): 1070–4. doi:10.1038/ejhg.2008.73. PMID 18364738.
- Oganesian A, Armstrong LC, Migliorini MM, Strickland DK, Bornstein P (February 2008). "Thrombospondins use the VLDL receptor and a nonapoptotic pathway to inhibit cell division in microvascular endothelial cells". Mol. Biol. Cell. 19 (2): 563–71. doi:10.1091/mbc.E07-07-0649. PMC 2230579. PMID 18032585.
- Wruss J, Rünzler D, Steiger C, Chiba P, Köhler G, Blaas D (May 2007). "Attachment of VLDL receptors to an icosahedral virus along the 5-fold symmetry axis: multiple binding modes evidenced by fluorescence correlation spectroscopy". Biochemistry. 46 (21): 6331–9. doi:10.1021/bi700262w. PMID 17472347.
- Suzuki K, Nakamura K, Iwata Y, Sekine Y, Kawai M, Sugihara G, Tsuchiya KJ, Suda S, Matsuzaki H, Takei N, Hashimoto K, Mori N (January 2008). "Decreased expression of reelin receptor VLDLR in peripheral lymphocytes of drug-naive schizophrenic patients". Schizophr. Res. 98 (1–3): 148–56. doi:10.1016/j.schres.2007.09.029. PMID 17936586. S2CID 45594329.
- Francis PJ, Hamon SC, Ott J, Weleber RG, Klein ML (May 2009). "Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss". J. Med. Genet. 46 (5): 300–7. doi:10.1136/jmg.2008.062737. PMID 19015224. S2CID 22940548.
- Zhang G, Assadi AH, McNeil RS, Beffert U, Wynshaw-Boris A, Herz J, Clark GD, D'Arcangelo G (2007). "The Pafah1b complex interacts with the reelin receptor VLDLR". PLOS ONE. 2 (2): e252. Bibcode:2007PLoSO...2..252Z. doi:10.1371/journal.pone.0000252. PMC 1800349. PMID 17330141.
- Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG (January 2008). "The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2". J. Biol. Chem. 283 (4): 2363–72. doi:10.1074/jbc.M708098200. PMID 18039658.
- Crawford DC, Nord AS, Badzioch MD, Ranchalis J, McKinstry LA, Ahearn M, Bertucci C, Shephard C, Wong M, Rieder MJ, Schellenberg GD, Nickerson DA, Heagerty PJ, Wijsman EM, Jarvik GP (March 2008). "A common VLDLR polymorphism interacts with APOE genotype in the prediction of carotid artery disease risk". J. Lipid Res. 49 (3): 588–96. doi:10.1194/jlr.M700409-JLR200. PMID 18056683.
- Yamada Y, Ando F, Shimokata H (July 2005). "Association of polymorphisms in CYP17A1, MTP, and VLDLR with bone mineral density in community-dwelling Japanese women and men". Genomics. 86 (1): 76–85. doi:10.1016/j.ygeno.2005.03.005. PMID 15953542.
- Chen Y, Hu Y, Lu K, Flannery JG, Ma JX (November 2007). "Very low density lipoprotein receptor, a negative regulator of the wnt signaling pathway and choroidal neovascularization". J. Biol. Chem. 282 (47): 34420–8. doi:10.1074/jbc.M611289200. PMID 17890782.
- Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA, Olson L, Kenealy SJ, Hauser M, Gilbert JR, Pericak-Vance MA (January 2006). "Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6". Invest. Ophthalmol. Vis. Sci. 47 (1): 329–35. doi:10.1167/iovs.05-0116. PMID 16384981.
- Sakai K, Tiebel O, Ljungberg MC, Sullivan M, Lee HJ, Terashima T, Li R, Kobayashi K, Lu HC, Chan L, Oka K (June 2009). "A neuronal VLDLR variant lacking the third complement-type repeat exhibits high capacity binding of apoE containing lipoproteins". Brain Res. 1276: 11–21. doi:10.1016/j.brainres.2009.04.030. PMC 2733343. PMID 19393635.
- Moser R, Snyers L, Wruss J, Angulo J, Peters H, Peters T, Blaas D (August 2005). "Neutralization of a common cold virus by concatemers of the third ligand binding module of the VLDL-receptor strongly depends on the number of modules". Virology. 338 (2): 259–69. doi:10.1016/j.virol.2005.05.016. PMID 15950998.
External links
- GeneReviews/NCBI/NIH/UW entry on VLDLR-Associated Cerebellar Hypoplasia or Dysequilibrium Syndrome-VLDLR
- Overview of all the structural information available in the PDB for UniProt: P98155 (Very low-density lipoprotein receptor) at the PDBe-KB.