Vitamin B12 total synthesis
The total synthesis of the complex biomolecule vitamin B12 was accomplished in two different approaches by the collaborating research groups of Robert Burns Woodward at Harvard[1][2][3][4][5] and Albert Eschenmoser at ETH[6][7][8][9][10][11][12] in 1972. The accomplishment required the effort of no less than 91 postdoctoral researchers (Harvard: 77, ETH: 14)[13]: 9-10 [14], and 12 Ph.D. students (at ETH[12]: 1420 ) from 19 different nations over a period of almost 12 years.[5]: 1:14:00-1:14:32,1:15:50-1:19:35 [14]: 17-18 The synthesis project[15] induced and involved a major change of paradigm[16][17]: 37 [18]: 1488 in the field of natural product synthesis.[19][20][21]
The molecule
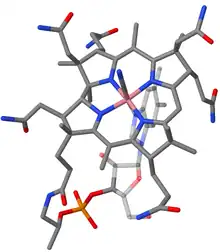
Vitamin B12, C63H88CoN14O14P, is the most complex of all known vitamins. Its chemical structure had been determined by x-ray crystal structure analysis in 1956 by the research group of Dorothy Hodgkin (Oxford University) in collaboration with Kenneth N. Trueblood at UCLA and John G. White at Princeton University.[24][25] Core of the molecule is the corrin structure, a nitrogenous tetradentate ligand system.[note 1] This is biogenetically related to porphyrins and chlorophylls, yet differs from them in important respects: the carbon skeleton lacks one of the four meso carbons between the five-membered rings, two rings (A and D, fig. 1) being directly connected by a carbon-carbon single bond. The corrin chromophore system is thus non-cyclic and expands over three meso positions only, incorporating three vinylogous amidine units. Lined up at the periphery of the macrocyclic ring are eight methyl groups and four propionic and three acetic acid side chains. Nine carbon atoms on the corrin periphery are chirogenic centers. The tetradentate, monobasic corrin ligand is equatorially coordinated with a trivalent cobalt ion which bears two additional axial ligands.[note 2]
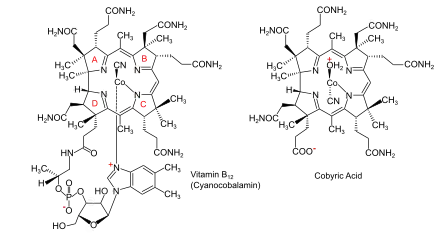
Several natural variants of the B12 structure exist that differ in these axial ligands. In the vitamin itself, the cobalt bears a cyano group on the top side of the corrin plane (cyanocobalamin), and a nucleotide loop on the other. This loop is connected on its other end to the peripheral propionic amide group at ring D and consists of structural elements derived from aminopropanol, phosphate, ribose, and 5,6-dimethylbenzimidazole. One of the nitrogen atoms of the imidazole ring is axially coordinated to the cobalt, the nucleotide loop thus forming a nineteen-membered ring. All side chain carboxyl groups are amides.
Cobyric acid, one of the natural derivatives of vitamin B12,[26] lacks the nucleotide loop; depending on the nature of the two axial ligands, it displays instead its propionic acid function at ring D as carboxylate (as shown in fig. 1), or carboxylic acid (with two cyanide ligands at cobalt).
The two syntheses
The structure of vitamin B12 was the first low-molecular weight natural product determined by x-ray analysis rather than by chemical degradation. Thus, while the structure of this novel type of complex biomolecule was established, its chemistry remained essentially unknown; exploration of this chemistry became one of the tasks of the vitamin's chemical synthesis.[12]: 1411 [18]: 1488-1489 [27]: 275 In the 1960s, synthesis of such an exceptionally complex and unique structure presented the major challenge at the frontier of research in organic natural product synthesis.[17]: 27-28 [1]: 519-521

Already in 1960, the research group of the biochemist Konrad Bernhauer in Stuttgart had reconstituted vitamin B12 from one of its naturally occurring derivatives, cobyric acid,[26] by stepwise construction of the vitamin's nucleotide loop.[note 4] This work amounted to a partial synthesis of vitamin B12 from a natural product containing all the structural elements of vitamin B12 except the nucleotide loop. Therefore, cobyric acid was chosen as the target molecule for a total synthesis of vitamin B12.[6]: 183-184 [1]: 521 [8]: 367-368
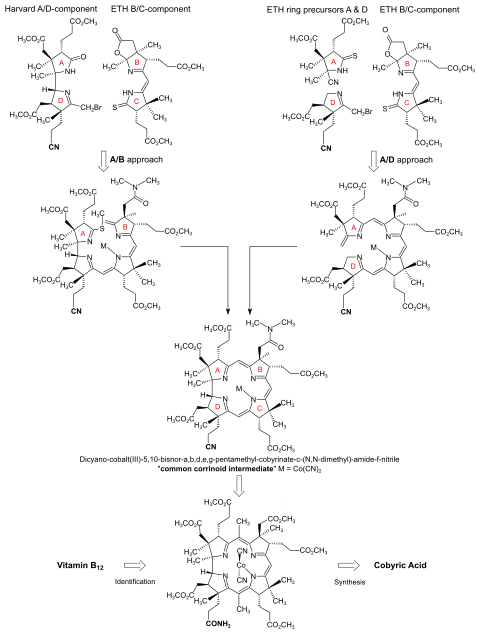
Collaborative work[3]: 1456 [17][30]: 302-313 of research groups at Harvard and at ETH resulted in two cobyric acid syntheses, both concomitantly accomplished in 1972,[31][32] one at Harvard[3], and the other at ETH.[10][11][12] A "competitive collaboration"[17]: 30 [33]: 626 of that size, involving 103 graduate students and postdoctoral researchers for a total almost 177 person-years,[13]: 9-10 is so far unique in the history of organic synthesis.[4]: 0:36:25-0:37:37 The two syntheses are intricately intertwined chemically,[18]: 1571 yet they differ basically in the way the central macrocyclic corrin ligand system is constructed. Both strategies are patterned after two model corrin syntheses developed at ETH.[8][18]: 1496,1499 [34]: 71-72 The first, published in 1964,[28] achieved the construction of the corrin chromophore by combining an A-D-component with a B-C-component via iminoester/enamine-C,C-condensations, the final corrin-ring closure being attained between rings A and B.[35] The second model synthesis, published 1969,[36] explored a novel photochemical cycloisomerization process to create the direct A/D-ring junction as final corrin-ring closure between rings A and D.[37]
The A/B approach to the cobyric acid syntheses was collaboratively pursued and accomplished in 1972 at Harvard. It combined a bicyclic Harvard A-D-component with an ETH B-C-component, and closed the macrocyclic corrin ring between rings A and B.[3]: 145,176 [4]: 0:36:25-0:37:37 The A/D approach to the synthesis, accomplished at ETH and finished at the same time as the A/B approach also in 1972, successively adds rings D and A to the B-C-component of the A/B approach and attains the corrin ring closure between rings A and D.[10][11][12] The paths of the two syntheses met in a common corrinoid intermediate.[11]: 519 [38]: 172 The final steps from this intermediate to cobyric acid were carried out in the two laboratories again collaboratively, each group working with material prepared via their own approach, respectively.[17]: 33 [18]: 1567
Synopsis of the Harvard/ETH collaboration
The beginnings
Woodward and Eschenmoser embarked on the project of a chemical synthesis of vitamin B12 independently from each other. The ETH group started with a model study on how to synthesize a corrin ligand system in December 1959.[18]: 1501 In August 1961,[17]: 29 [13]: 7 the Harvard group began attacking the buildup of the B12 structure directly by aiming at the most complex part of the B12 molecule, the "western half"[1]: 539 that contains the direct junction between rings A and D (the A-D-component). Already in October 1960,[17]: 29 [13]: 7 [39]: 67 the ETH group had commenced the synthesis of a ring-B precursor of vitamin B12.
At the beginning,[40] progress at Harvard was rapid, until an unexpected stereochemical course of a central ring formation step interrupted the project.[41][17]: 29 Woodward's recognition of the stereochemical enigma that came to light by the irritating behavior of one of his carefully planned synthetic steps became, according to his own writings,[41] part of the developments that led to the orbital symmetry rules.
After 1965, the Harvard group continued work towards an A-D-component along a modified plan, using (−)-camphor[42] as the source of ring D.[17]: 29 [18]: 1556
Joining forces: the A/B approach to cobyric acid synthesis
By 1964, the ETH group had accomplished the first corrin model synthesis,[28][27]: 275 and also the preparation of a ring-B precursor as part of a construction of the B12 molecule itself.[39][43] Since independent progress of the two groups towards their long-term objective was so clearly complementary, Woodward and Eschenmoser decided in 1965[18]: 1497 [17]: 30 to join forces and to pursue from then on the project of a B12 synthesis collaboratively, planning to utilize the ligand construction (ring coupling of components) strategy of the ETH model system.[2]: 283 [18]: 1555-1574
By 1966, the ETH group had succeeded in synthesizing the B-C-component ("eastern half"[1]: 539 ) by coupling their ring-B precursor to the ring-C precursor.[18]: 1557 The latter had also been prepared at Harvard from (−)-camphor by a strategy conceived and used earlier by A. Pelter and J. W. Cornforth in 1961.[note 6] At ETH, the synthesis of the B-C-component involved the implementation of the C,C-condensation reaction via sulfide contraction. This newly developed method turned out to provide a general solution to the problem of constructing the characteristic structural elements of the corrin chromophore, the vinylogous amidine systems bridging the four peripheral rings.[18]: 1499
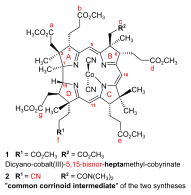
Early in 1967, the Harvard group accomplished the synthesis of the model A-D-component,[note 7] with the f-side chain undifferentiated, bearing a methyl ester function like all other side chains.[18]: 1557 From then on, the two groups systematically exchanged samples of their respective halves of the corrinoid target structure.[17]: 30-31 [18]: 1561 [32]: 17 By 1970, they had collaboratively connected Harvard's undifferentiated A-D-component with ETH's B-C-component, producing dicyano-cobalt(III)-5,15-bisnor-heptamethyl-cobyrinate 1 (fig. 4).[note 2] The ETH group identified this totally synthetic corrinoid intermediate by direct comparison with a sample produced from natural vitamin B12.[2]: 301-303 [18]: 1563
In this advanced model study, reaction conditions for the demanding processes of the C/D-coupling and the A/B-cyclization via sulfide contraction method were established. Those for the C/D-coupling were successfully explored in both laboratories, the superior conditions were those found at Harvard,[2]: 290-292 [18]: 1562 while the method for the A/B-ring closure via an intramolecular version of the sulfide contraction[46][36][47] was developed at ETH.[2]: 297-299 [48][18]: 1562-1564 Later it was shown at Harvard that the A/B-ring closure could also be achieved by thio-iminoester/enamine condensation.[2]: 299-300 [18]: 1564
By early 1971, the Harvard group had accomplished the synthesis of the final A-D-component,[note 8] containing the f-side chain carboxyl function at ring D differentiated from all the carboxyl functions as a nitrile group (as shown in 2 in fig. 4; see also fig. 3).[3]: 153-157 The A/D-part of the B12 structure incorporates the constitutionally and configurationally most intricate part of the vitamin molecule; its synthesis is regarded as the apotheosis of the Woodwardian art in natural product total synthesis.[11]: 519 [12]: 1413 [18]: 1564 [33]: 626
The alternative approach to cobyric acid synthesis
As far back as 1966,[37]: 1946 the ETH group had started to explore, once again in a model system, an alternative strategy of corrin synthesis in which the corrin ring would be closed between rings A and D. The project was inspired by the conceivable existence of a thus far unknown bond reorganisation process.[37]: 1943-1946 This – if existing – would make possible the construction of cobyric acid from one single starting material.[6]: 185 [8]: 392,394-395 [33] Importantly, the hypothetical process, being interpreted as implying two sequential rearrangements, was recognized to be formally covered by the new reactivity classifications of sigmatropic rearrangements and electrocyclizations propounded by Woodward and Hoffmann in the context of their orbital symmetry rules![8]: 395-397,399 [11]: 521 [49][18]: 1571-1572
By May 1968,[18]: 1555 the ETH group had demonstrated in a model study that the envisaged process, a photochemical A/D-seco-corrinate→corrinate cycloisomerization, does in fact exist. This process was first found to proceed with the Pd complex, but not at all with corresponding Ni(II)- or cobalt(III)-A/D-seco-corrinate complexes.[36][50]: 21-22 It also went smoothly in complexes of metal ions such as zinc and other photochemically inert and loosely bound metal ions.[8]: 400-404 [12]: 1414 These, after ring closure, could easily be replaced by cobalt.[8]: 404 These discoveries opened the door to what eventually became the photochemical A/D approach of cobyric acid synthesis.[7]: 31 [9]: 72-74 [37]: 1948-1959

Starting in fall of 1969[51]: 23 with the B-C-component of the A/B approach and a ring-D precursor prepared from the enantiomer of the starting material leading to the ring-B precursor, it took PhD student Walter Fuhrer[51] less than one and a half years[17]: 32 to translate the photochemical model corrin synthesis into a synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile 2 (fig. 4), the common corrinoid intermediate on the way to cobyric acid. At Harvard, the very same intermediate 2 was obtained around the same time by coupling the ring-D differentiated Harvard A-D-component (available in spring 1971[18]: 1564 footnote 54a [3]: 153-157 ) with the ETH B-C-component, applying the condensation methods developed earlier using the undifferentiated A-D-component.[1]: 544-547 [2]: 285-300
Thus, in spring 1971,[33]: 634 two different routes to a common corrinoid intermediate 2 (fig. 4) along the way to cobyric acid had become available, one requiring 62 chemical steps (Harvard/ETH A/B approach), the other 42 (ETH A/D approach). In both approaches, the four peripheral rings derived from enantiopure precursors possessing the correct sense of chiral, thereby circumventing major stereochemical problems in the buildup of the ligand system.[1]: 520-521 [7]: 12-13 [11]: 521-522 In the construction of the A/D-junction by the A/D-secocorrin→corrin cycloisomerization, formation of two A/D-diastereomers had to be expected. Using cadmium(II) as the coordinating metal ion led to a very high diastereoselectivity[51]: 44-46 in favor of the natural A/D-trans-isomer.[12]: 1414-1415
Once the corrin structure was formed by either approach, the three C-H-chirogenic centers at the periphery adjacent to the chromophore system turned out to be prone to epimerizations with exceptional ease.[2]: 286 [9]: 88 [3]: 158 [4]: 1:53:33-1:54:08 [18]: 1567 This required a separation of diastereomers after most of the chemical steps in this advanced stage of the syntheses. It was fortunate indeed that, just around that time, the technique of high pressure liquid chromatography (HPLC) had been developed in analytical chemistry.[52] HPLC became an indispensable tool in both laboratories;[32]: 25 [9]: 88-89 [3]: 165 [4]: 0:01:52-0:02:00,2:09:04-2:09:32 its use in the B12 project, pioneered by Jakob Schreiber at ETH,[53] was the earliest application of the technique in natural product synthesis.[18]: 1566-1567 [38]: 190 [54]
The joint final steps
The final conversion of the common corrinoid intermediate 2 (fig. 6) from the two approaches into the target cobyric acid required the introduction of the two missing methyl groups at the meso positions of the corrin chromophore between rings A/B and C/D, as well as the conversion of all peripheral carboxyl functions into their amide form, except the critical carboxyl at the ring-D f-side chain (see fig. 6). These steps were collaboratively explored in strictly parallel fashion in both laboratories, the Harvard group using material produced via the A/B approach, the ETH group such prepared by the photochemical A/D approach.[17]: 33 [18]: 1567
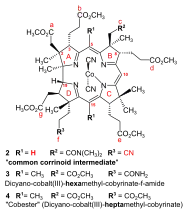
The first decisive identification of a totally synthetic intermediate on the way to cobyric acid was carried out in February 1972 with a crystalline sample of totally synthetic dicyano-cobalt(III)-hexamethyl-cobyrinate-f-amide 3 (fig. 6[note 2]), found to be identical in all data with a crystalline relay sample made from vitamin B12 by methanolysis to cobester 4,[note 9] followed by partial ammonolysis and separation of the resulting mixture.[55]: 44-45,126-143 [3]: 170 [57]: 46-47 At the time when Woodward announced the "Total Synthesis of Vitamin B12" at the IUPAC conference in New Delhi in February 1972,[3]: 177 the totally synthetic sample of the f-amide was one that had been made at ETH by the photochemical A/D approach,[17]: 35 [58]: 148 [18]: 1569-1570 while the first sample of synthetic cobyric acid, identified with natural cobyric acid, had been obtained at Harvard by partial synthesis from B12-derived f-amide relay material.[57]: 46-47 [3]: 171-176 Thus, the Woodward/Eschenmoser achievement around that time had been, strictly speaking, two formal total syntheses of cobyric acid, as well as two formal total syntheses of the vitamin.[57]: 46-47 [18]: 1569-1570
In the later course of 1972, two crystalline epimers of totally synthetic dicyano-cobalt(III)-hexamethyl-cobyrinate-f-amide 3, as well as two crystalline epimers of the totally synthetic f-nitrile, all prepared via both synthetic approaches, were stringently identified chromatographically and spectroscopically with the corresponding B12-derived substances.[18]: 1570-1571 [55]: 181-197,206-221 [5]: 0:21:13-0:46:32,0:51:45-0:52:49 [59] At Harvard, cobyric acid was then made also from totally synthetic f-amide 3 prepared via the A/B approach.[57]: 48-49 Finally, in 1976 at Harvard,[57] totally synthetic cobyric acid was converted into vitamin B12 via the pathway pioneered by Konrad Bernhauer.[note 4]
The publication record
Over the almost 12 years it took the two groups to reach their goal, both Woodward and Eschenmoser periodically reported on the stage of the collaborative project in lectures, some of them appearing in print. Woodward discussed the A/B approach in lectures published in 1968,[1] and 1971,[2] culminating in the announcement of the "Total Synthesis of Vitamin B12" in New Delhi in February 1972[3]: 177 published in 1973.[3] This publication, and lectures with the same title Woodward delivered in the later part of the year 1972[4][5] are confined to the A/B approach of the synthesis and do not discuss the ETH A/D approach.
Eschenmoser had discussed the ETH contributions to the A/B approach in 1968 at the 22nd Robert A. Welch Foundation conference in Houston,[7] as well as in his 1969 RSC Centenary Lecture "Roads to Corrins", published in 1970.[8] He presented the ETH photochemical A/D approach to the B12 synthesis at the 23rd IUPAC Congress in Boston in 1971.[9] The Zürich group announced the accomplishment of the synthesis of cobyric acid by the photochemical A/D-approach in two lectures delivered by PhD students Maag and Fuhrer at the Swiss Chemical Society Meeting in April 1972,[10] Eschenmoser presented a lecture "Total Synthesis of Vitamin B12: the Photochemical Route" for the first time as Wilson Baker Lecture at the University of Bristol, Bristol/UK on May 8, 1972.[note 10]


As a joint full publication of the syntheses by the Harvard and ETH groups (announced in[10] and expected in[11]) had not appeared by 1977,[note 12] an article describing the final version of the photochemical A/D approach already accomplished in 1972[10][51][55][63] was published 1977 in Science.[12][58]: 148 This article is an extended English translation of one that had already appeared 1974 in Naturwissenschaften,[11] based on a lecture given by Eschenmoser on January 21, 1974, at a meeting of the Zürcher Naturforschende Gesellschaft. Four decades later, in 2015, the same author finally published a series of six full papers describing the work of the ETH group on corrin synthesis.[64][18][65][66][35][37] Part I of the series contains a chapter entitled "The Final Phase of the Harvard/ETH Collaboration on the Synthesis of Vitamin B12",[18]: 1555-1574 in which the contributions of the ETH group to the collaborative work on the synthesis of vitamin B12 between 1965 and 1972 are recorded.
The entire ETH work is documented in full experimental detail in publicly accessible Ph.D. theses,[39][43][60][46][61][56][62][44][48][51][55][63] almost 1,900 pages, all in German.[67] Contributions of the 14 postdoctoral ETH researchers involved in the cobyric acid syntheses are mostly integrated in these theses.[12]: 1420 [64]: 1480 [13]: 12,38 The detailed experimental work at Harvard was documented in reports by the 77 postdoctoral researchers involved, with a total volume of more than 3,000 pages.[13]: 9,38 [note 11]
Representative reviews of the two approaches to the chemical synthesis of vitamin B12 have been published in detail by A. H. Jackson and K. M. Smith,[45] T. Goto,[68] R. V. Stevens,[38] K. C. Nicolaou & E. G. Sorensen,[15][19] summarized by J. Mulzer & D. Riether,[69] and G. W. Craig,[14][33] besides many other publications where these epochal syntheses are discussed.[note 13]
The Harvard/ETH approach to the synthesis of cobyric acid: the path to the common corrinoid intermediate via A/B-corrin-ring closure
In the A/B approach to cobyric acid, the Harvard A-D-component was coupled to the ETH B-C-component between rings D and C, and then closed to a corrin between rings A and B. Both these critical steps were accomplished by C,C-coupling via sulfide contraction, a new reaction type developed in the synthesis of the B-C-component at ETH. The A-D-component was synthesized at Harvard from a ring-A precursor (prepared from achiral starting materials), and a ring-D precursor prepared from (−)-camphor. A model A-D-component was used to explore the coupling conditions; this component differed from the A-D-component used in the final synthesis by having as the functional group at the ring-D f-side chain a methyl ester group (like all other side chains) instead of a nitrile group.
| The Harvard synthesis of the A-D-components for the A/B approach |
|---|
|
Synthesis of the ring-A precursor 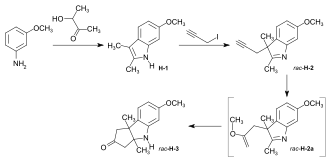 Figure 8: Harvard synthesis of the A-D-components: ring A Starting point for the synthesis of the ring-A precursor was methoxydimethyl-indol H-1 synthesized by condensation of the Schiff base from m-anisidine and acetoin. Reaction with the Grignard reagent of propargyl iodide gave racemic propargyl indolenine rac-H-2; ring closure to the aminoketone rac-H-3 was brought about by BF3 and HgO in MeOH through intermediate rac-H-2a (electrophilic addition) with the two methyl groups forced into a cis-relationship by kinetic as well as thermodynamic reasons.[1]: 521-522 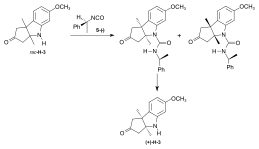 Figure 9: Harvard synthesis of the A-D-components: ring A resolution Resolution of the racemic aminoketone into the two enantiomers. Reaction of rac-H-3 with (−)-ethyl isocyanate permitted isolation by crystallization of one of the two diastereomeric urea derivatives formed (the other does not crystallize). Treatment of racemic ketone rac-H-3 (or of mother liquors from the previous crystallization) with (+)-ethyl isocyanate gave the enantiomer of the first urea derivative. Pyrolytic decomposition of each of these urea derivatives led to enantiopure aminoketones, the desired (+)-H-3, and (−)-H-3.[1]: 524-525 The "unnatural" (−)-enantiomer (−)-H-3 was used to determine the absolute configuration; in various later steps, (−)-H-3 and enantio-intermediates derived from it were used as model compounds in exploratory experiments.[38]: 173 Woodward wrote regarding the unnatural enantiomer "our experience has been such that this is just about the only kind of model study which we regard as wholly reliable".[1]: 529  Figure 10: Harvard synthesis of the A-D-components: Ring A determination of configuration Determination of the absolute configuration of ring-A precursor (+)-H-3. For this determination, the levo-rotatory ("unnatural") enantiomer of aminoketone (−)-H-3 was used in order to save precious material: Acylation of the amino group of (−)-H-3 with chloroacetyl chloride, followed by treatment of the product H-3a with potassium t-butoxide in t-butanol, afforded tetracyclic keto-lactame H-3b. Its keto carbonyl was converted to a methylene group by desulfurization of the dithioketal of H-3b with Raney nickel to give lactam H-3c. Destruction of the aromatic ring by ozonolysis, involving the loss of a carboxyl function by spontaneous decarboxylation, led to bicyclic lactam-carboxylic acid H-3d. This material was identified with a product H-3h derived from (+)-camphor, possessing the same constitution and the absolute configuration as shown in formula H-3d.[1]: 525-526 The material for this identification of H-3d was synthesized from (+)-camphor as follows: cis-isoketopinic acid H-3e, obtained from (+)-camphor by an established route described in the literature,[70] was converted via the correspondig chloride, azide, and isocyanate to methyl-urethane H-3f. When treated with potassium t-butoxide in t-butanol and subsequently with KOH, H-3f was converted to H-3h, clearly by way of the intermediate H-3g. The identity of the two samples of H-3d and H-3h obtained by the two routes described, established the absolute configuration of (+)-H-3, the enantiomer of the ring-A precursor.[1]: 525-526 Synthesis of the ring-D precursor from (-)-camphor 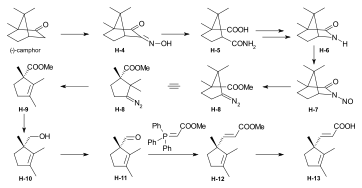 Figure 11: Harvard synthesis of the A-D-components: ring D from (-)-camphor (−)-Camphor was nitrosated in the α-position of the carbonyl group to give oxime H-4, Beckmann cleavage afforded via the corresponding nitrile the amide H-5. Hofmann degradation via an intermediary amine and its ring closure led to lactam H-6. Conversion of its N-nitroso derivative H-7 gave diazo compound H-8. Thermal decomposition of H-8 induced methyl migration to give cyclopentene H-9. Reduction to H-10 (LiAlH4), oxidation (chromic acid) to aldehyde H-11, Wittig reaction (carbomethoxymethylenetriphenylphosphorane) to H-12 and hydrolysis of the ester group finally gave trans-carboxylic acid H-13.[1]: 527-528 [note 14] Coupling of ring-A and ring-D precursors to "pentacyclenone" 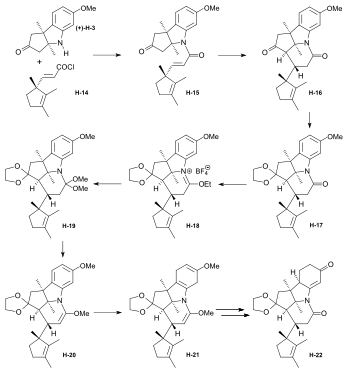 Figure 12: Harvard synthesis of the A-D-components: coupling of rings A and D to "pentacylenone" N-acylation of tricyclic aminoketone (+)-H-3 with the chloride H-14 of carboxylic acid H-13 gave amide H-15, which on treatment with potassium t-butoxide in t-butanol stereoselectively produced pentacyclic keto-lactam H-16 via an intramolecular Michael reaction which directs the indicated hydrogen atoms in trans relationship to each other. In anticipation of the Birch reduction of the aromatic ring, protective groups for the two carbonyl functions of H-16 were required, one for the ketone carbonyl group as ketal H-17, and the other for the lactam carbonyl as the highly sensitive enol ether H-20. The latter protection was achieved by treatment of H-17 with Meerwein salt (triethyloxonium tetrafluoroborate) to give iminium salt H-18, followed by conversion to orthoamide H-19 (NaOMe/MeOH), and finally expelling one molecule of methanol by heating in toluene. Birch reduction of H-20 (lithium in liquid ammonia, t-butanol, THF) provided tetraene H-21. Treatment with acid under carefully controlled conditions led first to an intermediate dione with the double bond in β,γ position which moved to the conjugated position in dione H-22, dubbed pentacyclenone.[1]: 528-531 [14]: 5 From "pentacyclenone" to "corrnorsterone" 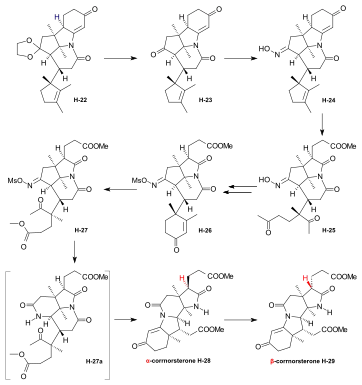 Figure 13: Harvard synthesis of the A-D-components: from "pentacylenone" to "corrnorsterone" The ethylene ketal protecting group in pentacyclenone H-22 was converted to the ketone group of H-23 by acid-catalyzed hydrolysis.[1]: 531 The dioxime primarily formed by reaction of diketone H-23 with hydroxylammonium chloride was regioselectively hydrolysed (nitrous acid/acetic acid) to the desired mono-oxime H-24. This is the oxime of the sterically more hindered ketone group, the nitrogen atom of which is destined to become the nitrogen of the target molecule's ring D. Crucial for this purpose is the configuration at the monoxime double bond, the hydroxyl group occupying the sterically less hindered position.[1]: 532 The C,C double bonds of both the cyclopentene and the cyclohexenone ring in H-24 were then cleaved by ozonolysis (ozone at 80 °C in MeOH, periodic acid), and the carboxylic group formed esterified with CH2N2) to diketone H-25. An intramolecular aldol condensation of the 1,5-dicarbonyl unit in MeOH using pyrrolidine acetate as the base, followed by tosylation of the oxime's hydroxyl group, afforded the cyclohexenone derivative H-26. A second ozonolysis in wet methyl acetate, followed by treatment with periodic acid and CH2N2 gave H-27. Beckmann rearrangement (MeOH, sodium polystyrene sulfonate, 2 hrs, 170 °C) produced regioselectively[1]: 532 lactam H-27a (not isolated) which reacted further in an amine-carbonyl condensation → aldol condensation cascade to the tetracycle H-28,[1]: 533-534 called α-corrnorsterone, implicating it as a "cornerstone"[1]: 534 in the synthesis of the desired A-D-component.[1]: 531-537 This compound required strongly alkaline conditions in order to open its lactam ring, but it was discovered that a minor isomer, also isolated from the reaction mixture, β-corrnorsterone H-29, undergoes this lactam ring opening under alkaline condition with great ease.[1]: 536 Structurally, the two isomers differ only in the orientation of the propionic acid side chain at ring A: the β-isomer has the more stable trans-orientation of this chain relative to the neighboring acetic acid chain formed after opening of the lactam ring. Equilibration of α-corrnorsterone H-28 by heating in strong base, followed by acidification and treatment with diazomethane, led to the isolation of pure β-corrnorsterone H-29 in 90 % yield.[1]: 537 The correct absolute configuration of the six contiguous asymmetric centers in β-corrnorsterone was confirmed by an x-ray crystal structure analysis of bromo-β-corrnorsterone[71][1]: 529 with the "unnatural" configuration.[1]: 538 [14]: 8 [4]: 0:49:20-0:50:42 Synthesis of the A-D-component carrying the propionic acid function at ring D as methoxycarbonyl group (model A-D-component)  Figure 14: Harvard synthesis of the A-D-components: f-undifferentiated model A-D-component Treatment of β-corrnorsterone H-29 with methanolic HCl cleaved the lactam ring and produced an enol ether derivative named hesperimine[note 15] H-30u. Ozonolysis to aldehyde H-32u, reduction of the aldehyde group with NaBH4 in MeOH to the primary alcohol H-33u and, finally, conversion of the hydroxy group via the corresponding mesylate gave bromide H-34u. This constitutes the model A-D-component, the one with an undifferentiated propionic acid function at ring D (i.e., bearing a methyl ester group like all other side chains).[1]: 539-540 Synthesis of the A-D-component carrying the propionic acid function at ring D as nitrile group 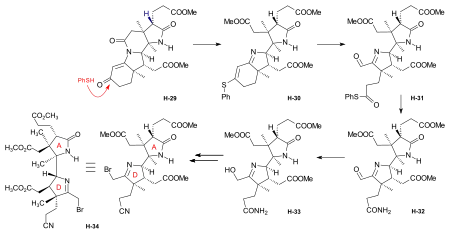 Figure 15: Harvard synthesis of the A-D-components: The f-differentiated A-D-component Conversion of β-corrnorsterone H-29 to the proper A-D-component H-34[1]: 538-539 containing the carboxyl function of the ring D propionic acid side chain as a nitrile group, differentiated from all the other methoxycarbonyl groups, involved the following steps: treatment of H-29 with a methanolic solution of thiophenol and HCl afforded phenyl-thioenolether derivative H-30, which upon ozonolysis at low temperature gave the corresponding thioester-aldehyde H-31 and, when followed by treatment with liquid ammonia, the amide H-32. Reduction of the aldehyde group with NaBH4 to H-33, mesylation of the primary hydroxy group with methanesulfonic anhydride under conditions that also convert the primary amide group into the desired nitrile group and, finally, replacement of the methansulfonyloxy group by bromide produced A-D-component H-34 with the propionic acid function at ring D as nitrile, differentiated from all other such side chains.[1]: 539-540 [4]: 1:01:56-1:19:47 |
| Coupling of Harvard A-D-components with the ETH B-C-component |
|---|
|
The construction of the corrin chromophore with its three vinylogous amidine units constitutes – besides the direct single bond connection between the rings A and D – the central challenge to any attempt to synthesize vitamin B12. The very first approach to a total synthesis of vitamin B12 launched by Cornforth[45]: 261-268 was discontinued when confronted with the task of coupling synthesized ring precursors.[18]: 1493,1496 Coupling the Harvard A-D-components with the ETH B-C-component required extensive exploratory work, this in spite of the knowledge gained in the ETH model syntheses of less complex (i.e., less peripherally substituted) corrins. What might be called an epic engagement for formally making just two C,C bonds lasted from early 1967[18]: 1557 until June 1970.[2] Both at ETH and Harvard, extensive model studies on the coupling of simplified enaminoid analogues of the A-D-component with the (ring C) imino- and thio-iminoester derivative of the full-fledged B-C-component had consistently shown that a coupling of the Harvard and the ETH components could hardly be achieved by the method that had been so successful in the synthesis of the simpler corrins, namely, by an intermolecular enamino-imino(or thio-imino)ester condensation[7][8][18]: 1561 [62]: 41-58 [1]: 544 [4]: 1:25:02-1:26:26 The outcome of these model studies determined the final structure type of a Harvard A-D-component: a structure capable of acting as a component of a C/D-coupling by sulfide contraction via alkylative coupling,[8]: 384-386 [47] i.e., the bromide H-34u.[7]: 18-22 [62]: 47,51-52 This method had already been implemented by the ETH group in the synthesis of the B-C-component.[33]: 16-19 [37]: 1927-1941 [18]: 1537-1540 An extensive search for optimal conditions, first for a C/D-coupling of a A-D-component with the ETH B-C-component E-19, then for conditions of the subsequent intramolecular A/B-corrin-ring closure was pursued in both laboratories, using the f-undifferentiated model A-D-component[note 7] H-34u[1]: 540 as a model.[2]: 287-300 [18]: 1561-1564 As the result of work by Yoshito Kishi at Harvard,[2]: 290 [18]: 1562 [14]: 11-12 and Peter Schneider at ETH,[48]: 12,22-29 [18]: 1563-1564 optimal conditions for the C/D-coupling were eventually found at Harvard, while the first and most reliable method for the corrin-ring closure between rings A and B was developed at ETH.[18]: 1562 The procedures of C/D-coupling and A/B-corrin-ring closure developed in this model series were later applied to the corresponding steps in the f-differentiated series as parts of the cobyric acid synthesis. Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,c,d,e,f,g-heptamethyl-cobyrinate from the ring-D undifferentiated model A-D-component D/C coupling.[7]: 22-23 [2]: 287-292 [48]: 12,22-28 [18]: 1561-1562  Figure 16: Harvard/ETH A/B approach to cobyric acid: D/C coupling of the Harvard model A-D-component with the ETH B-C-component The key problem in this step was the lability of the primary coupling product, thioether HE-35u, isomerizing to other thioethers at first not amenable to sulfide contraction in a reproducible procedure with acceptable yields.[2]: 287-290 [4]: 1:26:59-1:32:00 Induced by potassium t-butoxide in THF/t-butanol under rigorously controlled conditions with strict exclusion of air and moisture, the model A-D-component H-34u smoothly reacted with the B-C-component E-19[48]: 53-58 to give the sulfur-bridged coupling product HE-35u, named "thioether type I", in essentially quantitative yield.[2]: 287-288 However, this product could be isolated only under very carefully controlled conditions, since it equilibrates with extreme ease (e.g., chromatography, or traces of trifluoroacetic acid in methylenechloride solution) to the more stable isomeric thioether HE-36u (thioether type II) which contains, in contrast to thioether type I, the π-system of a conjugatively stabilized vinylogous amidine.[2]: 289 Depending on conditions, still another isomer HE-37u (thiother type III) was observed.[2]: 290 Starting with such mixtures of coupling products, at ETH a variety of conditions (e.g. methyl-mercury complex, BF3, triphenylphosphine[48]: 58-65 [2]: 291 ) were found to induce (via HE-38u) the contraction step to HE-39u in moderate yields.[18]: 1562 [2]: 287-292 With the choice of the solvent found to be crucial,[4]: 1:34:52-1:35:12 the optimal procedure at Harvard was heating thiother type II HE-36u in sulfolane in the presence of 5.3 equivalents trifluoroacetic acid and 4.5 equivalents of tris-(β-cyanoethyl)-phosphine at 60 °C for 20 hours, producing HE-39u in up to 85% yield.[2]: 292 [48]: 65-72 Later it was discovered that nitromethane could also be used as solvent.[4]: 1:34:52-1:35:13 [48]: 28 A/B-ring closure.[2]: 293-300 [48]: 12,29-39 [18]: 1562-1564 The problem of corrin-ring closure between rings A and B was solved in two different ways, one developed at ETH, the other pursued at Harvard.[32]: 19 Both methods correspond to procedures developed before in the synthesis of metal complexes[72] as well as free ligands[73] of simpler corrins.[7]: 25-28 [8]: 387-389 [18]: 1563 In the explorations of ring-closure procedures for the much more highly substituted A/B-seco-corrinoid intermediate HE-39u, the ETH group focused on the intramolecular version of the oxidative sulfide contraction method, eventually leading to the dicyano-cobalt(III)-complex HE48u.[48]: 29-39 [2]: 297-299 This first totally synthetic corrinoid intermediate was identified with a corresponding sample derived from vitamin B12.[18]: 1563 At Harvard, it was shown that the closure to the corrin macrocycle could also be realized by the method of thioiminoester/enamine condensation.[2]: 299-300 All reactions described here had to be executed on a very small scale, with "... the utmost rigour in the exclusion of oxygen from the reaction mixtures"[2]: 296 , and most of them also under strict exclusion of moisture and light, demanding very high standards of experimental expertise.[2]: 304 The major obstacle in achieving an A/B-corrin-ring closure was the exposure of the highly unstable ring B exocyclic methylidene double bond, which tends to isomerize into a more stable, unreactive endocyclic position with great ease.[48]: 86,97-98 [2]: 293-294 [3]: 161 [18]: 1562 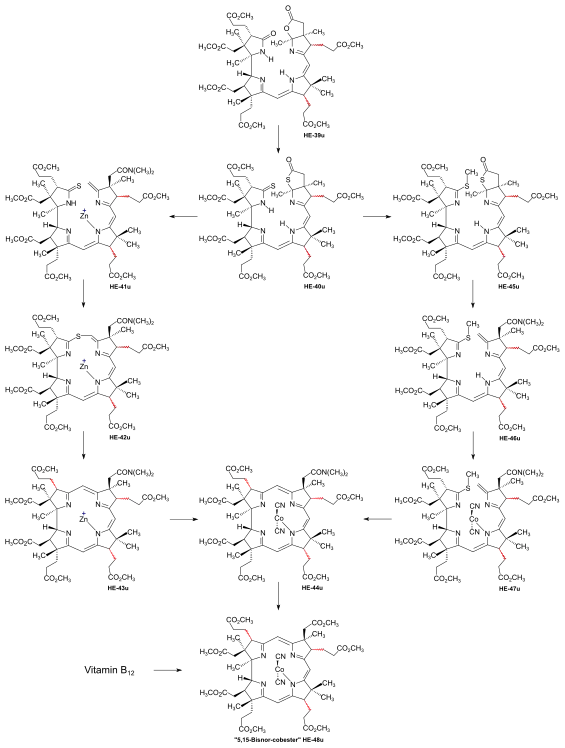 Figure 17: Harvard/ETH A/B approach to cobyric acid: A/B-ring closure to the f-undifferentiated model 5,15-bisnorcobyrinat The problem was solved at ETH[18]: 1562-1563 [48]: 29-39,126-135 by finding that treatment of the thiolactone-thiolactam intermediate HE-40u (obtained from HE-39u by reacting with P2S5[48]: 73-83 ) with dimethylamine in dry MeOH (room temperature, exclusion of air and light) smoothly opens the thiolactone ring at ring B, forming by elimination of H2S the exocyclic methylidene double bond as well as a dimethylamino-amide group in the acetic acid side chain.[48]: 32-34,96-99 These conditions are mild enough to prevent double bond tautomerization to the thermodynamically more stable isomeric position in the ring. Immediate conversion with a Zn-perchlorate-hexa(dimethylformamide) complex in methanol to zinc complex HE-41u, followed by oxidative coupling (0,05 mM solution of I2/KI in MeOH, 3 h) afforded HE-42u.[48]: 100-105 Sulfide contraction (triphenylphosphine, trifluoroacetic acid, 85 °C, exclusion of air and light) followed by re-complexation with Zn(ClO4)2 (KCl, MeOH, diisopropylamine) led to the chloro-zinc complex HE-43u.[48]: 105-116 The free corrinium salt formed when HE-43u was treated with trifluoroacetic acid in acetonitrile was re-complexed with anhydrous CoCl2 in THF to the dicyano-cobalt(III)-complex HE-44u.[48]: 117-125 [2]: 295 Conversion of the dimethylamino-amide group in the acetic acid side chain of ring B into the corresponding methylester group (O-methylation by trimethyloxonium tetrafluoroborate, followed by decomposition of the iminium salt with aqueous NaHCO3) afforded totally synthetic 5,15-bisnor-heptamethyl cobyrinate HE-48u.[48]: 11,117-125 A crystalline sample of HE-48u was identified via UV/VIS, IR, and ORD spectra with a corresponding crystalline sample derived from vitamin B12[48]: 42,135-141 [55]: 14,64-71,78-90 [2]: 287,301-303 [3]: 146-150 [74] Later at Harvard,[2]: 299-300 the A/B-corrin-ring closure was also achieved by converting the thiolactone-thiolactame intermediate HE-40u to thiolactone-thioiminoester HE-45u by S-methylation of the thiolactam sulfur (MeHgOi-Pr, then trimethyloxonium tetrafluoroborate). The product HE-45u was subjected to treatment with dimethylamine (as in the ETH variant), forming the highly labile methylidene derivative HE-46u, which then was converted with anhydrous CoCl2 in THF to dicyano-cobalt(III) complex HE-47u, the substrate ready to undergo the (A⇒B)-ring closure by a thioiminoester/enamine condensation. A careful search at Harvard for reaction conditions led to a procedure (KO-t-Bu, 120 °C, two weeks) that gave corrin Co complex HE-44u, identical with and in overall yields comparable with HE-44u obtained by the ETH variant of the sulfide contraction procedure.[2]: 300 Since in corrin model syntheses such a C,C-condensation required induction by a strong base, its application in a substrate containing seven methylester groups was not without problems;[18]: 1562 in a, milder reactions conditions were applied.[3]: 162 Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile (the common corrinoid intermediate) from the ring-D-differentiated A-D-component 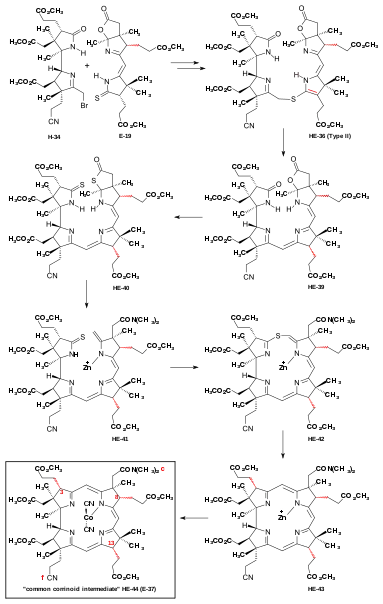 Figure 18: Harvard/ETH A/B approach to cobyric acid: coupling of the Harvard f-differentiated A-D-component with the ETH B-C-component to the common corrinoid intermediate The A-D-component H-34[note 8] with its propionic acid function at ring D differentiated from all the other carboxyl functions as nitrile group had become available at Harvard in spring 1971.[51]: 23 As a result of the comprehensive exploratory work that had been done with the model A-D-component at Harvard and ETH,[2]: 288-292 [48]: 22-28 [18]: 1561-1562 joining the proper A-D-component H-34 with the B-C-component E-19 by three operations H-34 + E-19 →→ HE-36 → HE-39.[3]: 158-159 [4]: 1:19:48-1:36:15 Closing the corrin ring was achieved in the sequence HE-39 (P2S5, xylene, γ-picoline)→ HE-40[4]: 1:36:45-1:37:49 → HE-41[4]: 1:37:51-1:42:33 → HE-42[4]: 1:42:35-1:44:34 → HE-43 (overall yield "about 60 %"[4]: 1:44:35-1:46:32 ), and finally to cobalt complex HE-44.[4]: 1:46:34-1:52:51 [3]: 160-166 Reactions in this sequence were based on the procedures developed in the undifferentiated model series.[2]: 293-300 [48]: 29-39 [18]: 1562-1564 Two methods were available for the A/B-ring closure: oxidative sulfide contraction within a zinc complex, followed by exchange of zinc by cobalt (ETH[3]: 162-165 ), or the Harvard alkylative variant of a sulfide contraction,[3]: 160-162 thio-iminoester/enamine condensation of the cobalt complex (improved reaction conditions: diazabicyclononanone in DMF, 60 °C, several hours[3]: 162 ). Woodward preferred the former one:[3]: 165 "...the oxidative method is somewhat superior, in that it is relatively easier to reproduce, .... ".[4]: 1:52:37-1:53:06 The corrin complex dicyano-cobalt(III)-5,15-bisnor-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile HE-44 took up the role of the common corrinoid intermediate in the two approaches to cobyric acid synthesis: HE-44 ≡ E-37. Due to the high configurational lability of C-H chirogenic centers C-3, C-8 and C-13[4]: 1:21:49-1:23:42,1:35:43-1:36:14,1:51:51-1:52:30 at the ligand periphery in basic or acidic milieu, separation by HPLC was indispensable for isolation, purification and characterization of pure diastereomers of this and the following corrinoid intermediates.[3]: 165-166 [9]: 88-89 [4]: 1:53:07-2:01:24 |
| Preparation of ring-C precursor from (+)-camphor by the Harvard group |
|---|
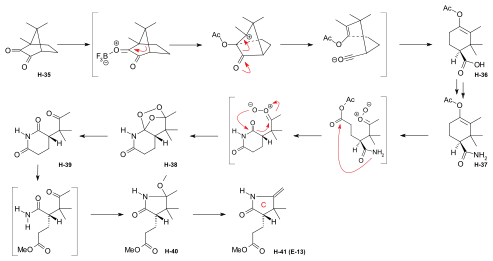 Figure 19: Harvard preparation of the ring-C precursor from (+)-camphor Starting material for the synthesis of a ring-C precursor was (+)-camphorquinone H-35[note 16] which was converted to the acetoxy-trimethylcyclohexene-carboxylic acid H-36 by BF3 in acetic anhydride, a reaction pioneered by Manasse & Samuel in 1902,[75], already successfully applied in a previous synthesis of the ring-C precursor by Pelter and Cornforth.[note 6] Conversion of H-36 to amide H-37 was followed by its ozonolysis to peroxide H-38 which was reduced to the keto-succinimide H-46 by zinc and MeOH. Treatment with methanolic HCl gave lactam H-40, followed by thermal elimination of methanol to the ring-C precursor H-41[1]: 540-542 [48]: 49-50 [14]: 4-5,15 This was found to be identical with the ring-C precursor E-13 prepared by a different route[note 5] at ETH.[61]: 32 [44]: 30,33-34,81 |
The ETH approach to the synthesis of cobyric acid: the path to the common corrinoid intermediate via A/D-corrin-ring closure
In the A/D approach to the synthesis of cobyric acid, the four ring precursors (ring-C precursor only formally so[12]: ref. 22 ) derive from the two enantiomers of one common chiral starting material. All three vinylogous amidine bridges that connect the four peripheral rings were constructed by the sulfide contraction method, with the B-C-component – already prepared for the A/B-approach – serving as an intermediate.[12][11] The photochemical A/D-secocorrin→corrin cycloisomerization, by which the corrin ring was closed between rings A and D, is a novel process, targeted and found to exist in a model study (cf. fig. 2).[36][37]: 1943-1948
| Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach) |
|---|
|
Syntheses of the ring-B precursor Two syntheses of ring-B precursor (+)-E-5 were realized; the one starting from 2-butanone was used further.[6]: 188 Two pathways for the conversion of the ring-B precursor into the ring-C precursor (+)-E-5 → (−)-E-13 ≡ H-41 were developed, one at ETH,[44]: 15-39 [1]: 544 , and one at Harvard.[6]: 193 [note 17] These conversions turned out to be inadequate for producing large amounts of ring-C-precursor.[46]: 38 [18]: 1561 However, the pathway developed at ETH served the purpose of determining the absolute configuration of the ring-B precursor.[6]: 193 [61]: 32 Bulk amounts of ring-C precursor to be used for the production of the B-C-component at ETH[44]: 40 [6]: 193 [33]: 631 were prepared at Harvard from (+)-camphor by a route originally developed by Pelter and Cornforth.[note 6] 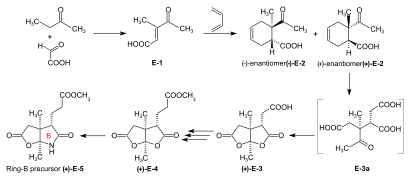 Figure 20: ETH synthesis of the B-C-component: synthesis of the two enantiomers of the ring-B precursor Ring-B precursor from 2-butanone and glyoxylic acid. Aldol condensation between 2-butanone and glyoxylic acid by treatment with concentrated phosphoric acid) gave stereoselectively (trans)-3-methyl-4-oxo-2-pentenoic acid E-1.[39]: 11-20,45-45 Diels-Alder reaction of E-1 with butadiene in benzene in the presence of SnCl4 afforded the racemate of the chiral Diels-Alder adduct E-2 which was resolved into the enantiomers by sequential salt formation with both (−)- and (+)-1-phenylethylamine.[43]: 22,59-62 The chirogenic centers of the (+)-enantiomer (+)-E-2 possessed the absolute configuration of ring B in vitamin B12.[60]: 35 [6]: 191 Oxidation of this (+)-enantiomer with chromic acid in acetone in the presence of sulfuric acid afforded the dilactone (+)-E-3 of the intermediary tricarboxylic acid E-3a.[43]: 35,72-73 Thermodynamic control of dilactone formation leads to the cis-configuration of the ring junction.[43]: 32-34 Elongation of the acetic acid side chain of (+)-E-3 by the Arndt-Eistert reaction (via the corresponding acid chloride and diazoketone) gave dilactone (+)-E-4.[61]: 15-16,65-67 Treatment of (+)-E-4 with NH3 in MeOH at room temperature formed a dual mixture of isomeric lactam-lactones in a ratio of 2:1, with ring-B precursor (+)-E-5 predominating (isolated in 55% yield).[46]: 12-17,57-63 [6]: 186-188 [12][1]: 542-543 The isomeric lactam-lactone could be isomerized to (+)-E-5 by treatment in methanolic HCl.[61]: 24-26,81-84 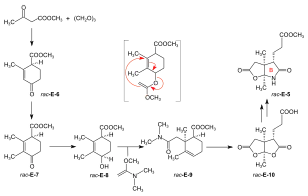 Figure 21: ETH synthesis of the B-C-component: Alternative Synthesis of the (racemic) Ring-B Precursor (only one enantiomer shown for racemates) Alternative synthesis of racemic ring-B precursor from Hagemann's ester: implementation of the amidacetal-Claisen rearrangement. Five steps were needed to transform Hagemann's ester rac-E-6 into the racemate of the lactam-lactone rac-E-5 form of the ring-B precursor.[60]: 14-31 [6]: 188-190 The product of the C-methylation step rac-E-6 → rac-E-7 (NaH, CH3I) was purified via its crystalline oxime. The cis-hydroxy-ester (configuration secured by lactone formation[60]: 64 ) resulting from the reduction step rac-E-7 → rac-E-8 (NaBH4) had to be separated from the trans isomer. The thermal rearrangement rac-E-8 → rac-E-9 constitutes the implementation of the amidacetal-Claisen rearrangement in organic synthesis,[76][60]: 36-49 a precedent to Johnson's orthoester-Claisen and Ireland's ester-enolate rearrangement.[77] Ozonolysis (O3/MeOH, HCOOH/H2O2) of the N,N-dimethylamide ester rac-E-9 afforded dilactone acid rac-E-10, from which two reactions led to lactam-lactone methylester rac-E-7, the racemate of ring-B precursor (+)-E-7.[60]: 57-67 Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor 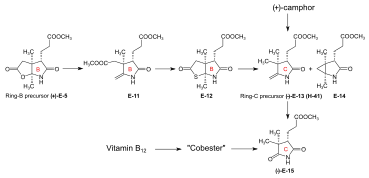 Figure 22: ETH synthesis of the B-C-component: Conversion of Ring-B Precursor to Ring-C Precursor The conversion of ring-B precursor into the ring-C precursor was based on a reductive decarbonylation of thiolactone E-12 with chloro-tris-(triphenylphosphino)-rhodium(I).[44]: 14-32 [6]: 191-193 [12] Treatment of a methanolic solution of ring-B precursor (+)-E-5 with diazomethane in the presence of catalytic amounts of sodium methoxide, followed by thermal elimination of methanol, gave methylidene lactam E-11, which was converted to the thiolactone E-12 with liquid H2S containing a catalytic amount of trifluoracetic acid.[44]: 15-16,56-58 Heating E-12 in toluene with the Rh(I)-complex afforded ring-C precursor (−)-E-13 besides the corresponding cyclopropane derivative E-14. Ring-C precursors prepared via this route and from (+)-camphor at Harvard [1]: 540-542 were found to be identical: (−)-E-13 ≡ H-41.[44]: 33-34 Ozonolysis of ring-C precursor (−)-E-13 gave succinimide derivative (−)-E-15.[44]: 33-35,88-89 This succinimide was found to be identical[6]: 193 [1]: 543-544 in constitution and optical rotation (i.e., configuration) with the corresponding succinimide derived from ring C of Vitamin B12, isolated after ozonolysis of crystalline heptamethyl-cobyrinate (cobester[note 9]) prepared from Vitamin B12.[56]: 9-18,67-70 The approach pursued at Harvard for conversion of ring-B precursor into ring-C precursor was based on a photochemical degradation of the acetic acid side chain carboxyl group, starting from (+)-E-7 prepared at ETH.[note 17] Coupling of ring-B and ring-C precursors to the B-C-component. Implementation of the sulfide contraction C,C-condensation method The iminoester/enamine C,C-condensation method for constructing the vinylogous amidine system, developed in the model studies on corrin synthesis,[28][35] failed completely in attempts to create the targeted C,C-bond between ring-B precursor (+)-E-5 with ring-C precursor (−)-E-13 to give the B-C-component E-18.[6]: 193-194 [8]: 379 [1]: 544 The problem was solved by "intramolecularization" of the bond formation process between the electrophilic (thio)iminoester carbon and the nucleophilic methylidene carbon of the enamine system through first oxidatively connecting these two centers by a sulfur bridge, and then achieving the C,C-bond formation by a now intramolecular thio-iminoester/enamine condensation with concomitant transfer of the sulfur to a thiophile.[6]: 194-197 [8]: 380-386 [18]: 1537-1538 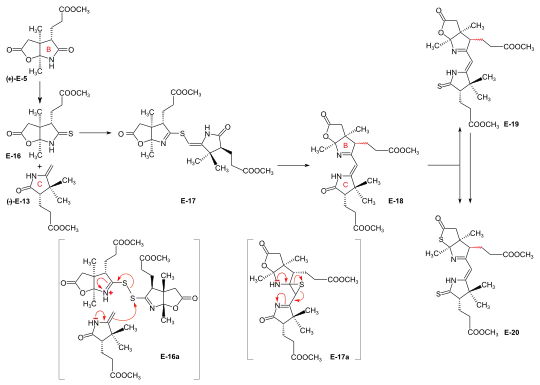 Figure 23: ETH synthesis of the B-C-component: coupling of the ring B and C precursors (implementation of C/C-coupling by the sulfide-contraction method) Conversion of lactam (+)-E-5 into the corresponding thiolactam E-16 (P2S5),[46]: 20-23,74-75 oxidation of E-16 with benzoyl peroxide in the presence of ring-C precursor (−)-E-13 (prepared at Harvard by the Cornforth route[note 6]), followed by heating the reaction product E-17 in triethylphosphite (as both solvent and thiophile) afforded B-C-component E-18 as a (not separated) mixture of two epimers (regarding the configuration of the propionic side chain at ring B) in up to 80 % yield.[46]: 38-43,96-102 [33]: 16-19 [8]: 381-383 [48]: 20-21,50-52 The bracketed formulae in the reaction scheme illustrate the type of mechanism operating in the process: E-16a = primary coupling of E-12 and E-10 to E-13; E-17a = extrusion of the sulfur atom (captured by thiophile) to E-14, where it is left open whether this latter process occurs at the stage of the episulfide. This reaction concept developed at this stage, dubbed sulfide contraction,[6]: 199 [47][18]: 1534-1541 [37]: 1927-1941 turned out to make possible the construction of all three meso-carbon bridges of the vitamin's corrin ligand in both approaches of the synthesis.[12][11][2]: 288-292,297-300 [3]: 158-164 The conversion of bicyclic lactone-lactam E-18 into the corresponding thiolactone-thiolactam E-20 was brought about by heating with P2S5/4-methylpyridine in xylene at 130 °C; milder condition produced thiolactam-lactone E-19, used for coupling with the Harvard A-D-components.[51]: 73-83 |
| Coupling of the B-C-component with ring-D and ring-A precursors |
|---|
|
Synthesis of ring-D precursor for the A/D approach 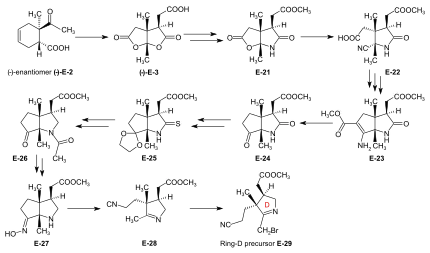 Figure 24: ETH A/D approach to cobyric acid: synthesis of ring-D precursor The starting material for the ring-D precursor,[61]: 40-61 [63]: 17-22 [12] the (−)-enantiomer of the dilactone-carboxylic acid (−)-E-3, was prepared from the (−)-enantiomer of the Diels-Alder adduct (−)-E-2[note 18] by oxydation with chromic acid/sulfuric acid in acetone.[43]: 35,72-73 Treatment of (−)-E-3 with NH3 in MeOH gave a lactone-lactam-acid which was esterified with diazomethane to the ester E-21,[61]: 104-110 the lactone ring of which was opened with KCN in MeOH to give E-22.[61]: 114-116 Conventional conditions of an Arndt-Eistert reaction (SOCl2: acid chloride, then CH2N2 in THF: diazoketone, treated with Ag2O in MeOH) led to an – unforeseen, yet useful – ring closure of the originally formed chain-elongated ester through participation of the cyano group as a neighboring electrophile, affording the bicyclic enamino-ester derivative E-23.[61]: 116-120 Hydrolysis with aqueous HCl, accompanied by decarboxylation, and re-esterification with diazomethane gave keto-lactam-ester E-24.[61]: 123-126 [63]: 40-41 Ketalization ((CH2OH)2, CH(OCH3)3, TsOH) of E-24 and conversion of this lactam-ester to thiolactam E-25 (P2S5) was followed by reductive removal of the sulfur with Raney nickel, acetylation of the amino group, and hydrolysis of the ketal (AcOH) to afford E-26.[63]: 42-59 This was converted by deacetylation of the amino group with HCl, and then by treatment with NH2OH/HCl, MeOH/NaOAc into oxime E-27. Beckmann fragmentation (HCl, SOCl2 in CHCl3, N-polystyryl-piperidine) of this oxime E-27 produced imino-nitrile E-28,[63]: 60-67 which, when treated with bromine (in MeOH, phosphate buffer pH 7.5, -10 °C) gave ring-D precursor E-29.[51]: 84-88 Conversion of the ring-B precursor into the ring-A precursor for the A/D approach  Figure 25: ETH A/D approach to cobyric acid: conversion of ring-B precursor into ring-A precursor The ring-A precursor (−)-E-31 required in the A/D approach is a close derivative of ring-B precursor (+)-E-5. Its preparation from (+)-E-5 required opening of the lactone group (KCN in MeOH), followed by re-esterification with diazomethane to E-30, then conversion of the lactam group into a thiolactam group with P2S5 to yield (−)-E-31.[51]: 63-72 [12] Coupling of the B-C-component with ring-D and ring-A precursors The most efficient way of attaching the two rings D and A to the B-C-component E-18 was to convert E-18 directly into its thiolactam-thiolactone derivative E-20 and then to proceed by first coupling ring-D precursor E-29 to ring C, and then ring-A precursor E-31 to ring B, both by the sulfide contraction method.[51]: 26-31 [9]: 80-83 [12] The search for the reaction conditions for these attachments was greatly facilitated by exploratory work done on the two sulfide contraction steps in the A/B approach model study.[51]: 27 [48]: 22-39 [2]: 285-300 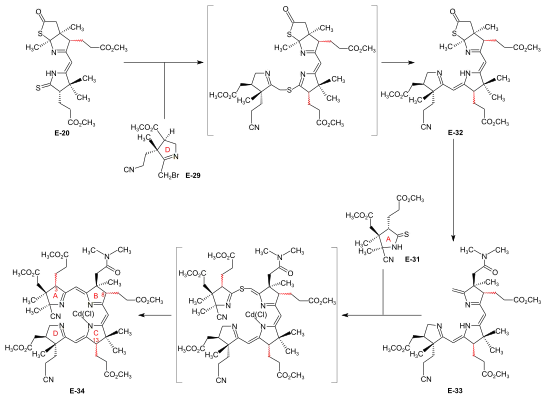 Figure 26: ETH A/D approach to cobyric acid: Attaching ring-C and ring-A precursors to the B-C-component to yield the A/D-seco-corrin Attachment of ring-D precursor E-29 to the ring-C thiolactam in E-20 by sulfide contraction via alkylative coupling (t-BuOK in t-BuOH/THF, tris-(β-cyano-ethyl)-phosphin/CF3COOH in sulfolane) afforded the B/C/D-sesqui-corrinoid E-32.[51]: 89-97 To attach ring-A precursor E-31, the ring B of E-32 was induced to expose its exocyclic methylidene double bond by treatment with dimethylamine in MeOH (using the method[note 19] developed by Schneider[48]: 32-34 ) forming E-33[51]: 108-115 which was subjected to the following cascade of operations:[51]: 130-150 iodination (N-iodosuccinimide, CH2Cl2, 0°), coupling with the thiolactam sulfur of the ring-A precursor E-31 [(CH3)3Si]2N-Na in benzene/t-BuOH), complexation (Cd(ClO4)2 in MeOH), treatment with triphenylphosphine/CF3COOH in boiling benzene (sulfide contraction) and, finally, re-complexation with Cd(ClO4)2/N,N-diisopropylethylamine in benzene/MeOH). These six operations, all carried out without isolation of intermediates, gave A/D-seco-corrin complex E-34 as mixture of peripheral epimers (separable via HPLC[51]: 143-147 ) in 42-46 % overall yield.[51]: 139 |
| A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization |
|---|
|
A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization to dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile (the common corrinoid intermediate) The conditions and prerequisites for the final (A⇒D)-corrin-ring closure were taken over from extensive corrin model studies.[36][78][9]: 71-74,83-84 [18]: 1565-1566 [37]: 1942-1962 Problems specific to the cobyric acid synthesis that had to be tackled were:[9]: 84-88 the possible formation of two diastereomeric A/D-trans-junctions in the ring closure,[51]: 37-38 exposure of the methylidene double bond at ring A of the A/D-seco-corrin E-34 in a labile Cd complex,[51]: 35-36 [18]: 1566 and epimerizability of the peripheral stereogenic centers C-3, C-8 and C-13 before and after ring closure.[51]: 39 [3]: 148-150 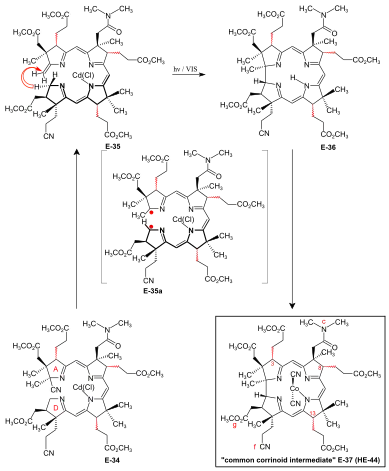 Figure 27: ETH A/D approach to cobyric acid: photochemical A/D-seco-corrin→corrin cycloisomerization to the common corrinoid intermediate 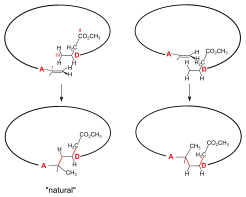 Figure 28: ETH A/D approach to cobyric acid: coil selectivity in A/D-ring closure In the application of this novel process in the A/D approach of the cobyric acid synthesis,[9]: 86-95 [51]: 39-53 [12]: 1419 the reaction proceeded most efficiently and with highest coil stereoselectivity in favor of the natural A/D-trans junction in an A/D-seco-corrin cadmium complex.[51]: 42-45 [3]: 166 Treatment of Cd-complex E-34 as mixture of peripheral epimers with 1,8-Diazabicyclo(5.4.0)undec-7-ene in sulfolane at 60 °C under strict protection against light to eliminate the cyano group at ring A, directly followed by re-treatment with Cd(ClO4)2, led to labile[51]: 172 A/D-seco-corrin complex E-35 as a mixture of peripheral epimers. This was directly subjected to the key step, the photochemical ring closure reaction under rigorous exclusion of air:[51]: 40 visible light, under Argon, MeOH, AcOH, 60 °C. Product of the A/D-ring closure was the free corrin ligand E-36, as the originally formed Cd-corrinate – in contrast to the Cd-seco-corrinate E-35 – decomplexes in the reaction medium.[51]: 173 [12]: 1419 Corrin E-36 was immediately complexed (CoCl2,[18]: 1499-1500,1563-64 KCN, air, H2O, CH2Cl2) and finally isolated (thick-layer chromatography) as mixture of peripheral epimers in 45-50 % yield over four operations:[51]: 169-179 the common corrinoid intermediate dicyano-cobalt(III)-complex E-37 ≡ HE-44.[note 20] HPLC analysis of this mixture E-37 showed the presence of six epimers with natural ligand helicity (Σ 95%, CD spectra), among them 26% of natural diastereomer 3α,8α,13α, and an equal amount of its C-13 neo-epimer 3α,8α,13β.[51]: 46,179-186 [12]: 1414 Two HPLC fractions (Σ 5%) contained diastereomers with unnatural ligand helicity, as shown by inverse CD spectra.[51]: 42-43 Product mixtures from several such cycloisomerizations were combined for preparative HPLC separation and full characterization of the 14 isolated diastereomers of E-37[51]: 207-251 (of 16 theoretically possible, regarding helicity and the epimeric centers C-3, C-8, C-13[51]: 39 ). In an analytical run, the mixture of cadmium-seco-complex epimers E-35 was separated by HPLC (in the dark) into the natural chloro-cadmium-3α,8α,13α-A/D-seco-corrinate diastereomer (ααα)-E-35 and four other epimer fractions[51]: 281-293 Upon irradiation[51]: 53 [12] and following cobaltation, (ααα)-E-35 produced E-37 in yields of 70-80% as an essentially dual mixture of mainly the 3α,8α,13α epimer, besides some 3α,8α,13β epimer. Less than 1% of fractions with unnatural coil were formed (HPLC, UV/VIS, CD).[51]: 293-300 Mechanistically, the photochemical A/D-seco-corrin corrin cycloisomerization involves an antarafacial sigmatropic shift of the α-hydrogen of the CH2 position C-19 at ring D to the CH2 position of the methylidene group at ring A within a triplet excited state, creating a transient 15-center-16-electron π-system (see E-35a in fig. 27) that antarafacially collapses between positions C-1 and C-19 to the corrin system.[36][37]: 1946,1967-1993 [79] The coil selectivity of the ring closure in favor of the corrin ligand's natural helicity is interpreted as relating to the difference in steric hindrance between the g-methoxycarbonyl acetic acid chain at ring D and the methylidene region of ring A in the two possible helical coil configurations of the A/D-seco-corrin complex (fig. 28).[51]: 38 [37]: 1960-1962 |
ETH/Harvard: the jointly executed final steps from the common corrinoid intermediate to cobyric acid
The final steps from the common corrinoid intermediate E-37/HE-44 to cobyric acid E-44/HE-51 were carried out by the two groups collaboratively and in parallel, the ETH group working with material produced by the A/D approach, and the Harvard group with that from the A/B approach.[63]: 15 [55]: 22 [57]: 47 [14]: 12 [18]: 1570-1571 What the two groups in fact accomplished thus were the common final steps of two different syntheses.[11][12]
The tasks in this end phase of the project were the regioselective introduction of methyl groups at the two meso positions C-5 and C-15 of E-37/HE-44, followed by conversion of all its peripheral carboxyl functions into primary amide groups, excepting that in side chain f at ring D, which had to end up as free carboxyl. These conceptually simple finishing steps turned out to be rather complex in execution, including unforeseen pitfalls like a dramatic loss of precious synthetic material in the so-called "Black Friday" (July 9, 1971).[55]: 39-40,107-118 [9]: 97-99 [3]: 168-169 [5]: 0:07:54-0:09:33 [18]: 1568-1569
| Introduction of methyl groups in two meso positions |
|---|
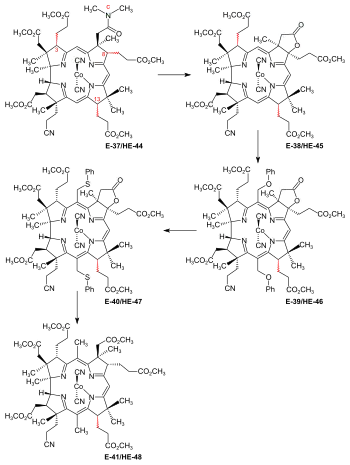 Figure 29: ETH/Harvard joint final steps: Introduction of methyl groups at the meso positions C-5 and C-15 This introduction of methyl groups could draw on exploratory studies on model corrins[7]: 13-14 [8]: 375-377 [80][18]: 1528,1530-1532 as well as on exploratory experiments carried out at ETH on cobester[note 9] and its (c→C-8)-lactone derivative.[55]: 27-43 Chloromethyl benzyl ether alkylated the meso position C-10 of cobester, but not that of the corresponding lactone, the difference in behavior reflecting the difference in steric hindrance exerted on the meso position C-10 by its neighboring substituents.[55]: 37-39 This finding was decisive for the choice of the substrate to be used for introducing methyl groups at meso positions C-5 and C-10 of E-37/HE-44.[9]: 96-99 [55]: 19 [3]: 167 [18]: 1567-1568 In this final phase of the synthesis, HPLC again turned out to be absolutely indispensable for separation, isolation, characterization and, above all, identification of pure isomers of dicyano-cobalt(III)-complexes of totally as well as partially synthetic origin.[9]: 96-102 [3]: 165 [55]: 61-63 [5]: 0:21:13-0:25:28 [18]: 1566-1567 The first step was to convert the c-N,N-dimethylcarboxamide group of E-37/HE-44 into the (c→C-8)-lactone derivative E-38/HE-45 by treatment with iodine/AcOH effecting iodination at C-8, followed by intramolecular O-alkylation of the carboxamide group to an iminium salt that hydrolyses to the lactone.[63]: 23,90-108 [3]: 166-167 [4]: 2:02:18-2:09:02 This lactonization leads to cis-fused rings.[55]: 19 [5]: 0:09:34-0:10:43 Reaction of (c→C-8)-lactone E-38/HE-45 with chloromethyl benzyl ether in acetonitrile in the presence of LiCl gave, besides mono-adduct, the bis-benzyloxy adduct E-39/HE-46. When treated with thiophenol, this produced the bis-phenylthio-derivative E-40/HE-47. Treatment with Raney nickel in MeOH not only set free the two methyl groups at the meso positions, but also reductively opened the lactone ring to the free c-carboxyl group at ring B, producing the correct α-configuration at C-8. Esterification of c-carboxyl with diazomethane afforded hexamethylester-f-nitrile E-41/HE-48.[55]: 19-21,39-43,146-205 [3]: 167-169 For steric reasons, only the predominant[55]: 19 [63]: 24 [4]: 2:08:20-2:09:02 C-3 α-epimer (with the C-3 side chain below the plane of the corrin ring) reacted to a 5,15-disubstituted product E-38/H-45, the reaction thus amounting to a chemical separation of the C-3 epimers.[55]: 40 [5]: 0:12:51-0:14:33,0:15:56-0:16:24 In improved procedures developed at Harvard later in 1972,[18]: 1569 footnote 62 the reagent chloromethyl benzyl ether was replaced by formaldehyde/sulfolane/HCl in acetonitrile for the alkylation step, and Raney nickel in the reduction step was replaced by zinc/acetic acid to give E-41/HE-48.[5]: 0:00:32-0:21:12 |
| Dicyano-cobalt(III)-3α,8α,13α-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide: Identification with material derived from vitamin B12 |
|---|
|
Concentrated H2SO4 at room temperature converted the nitrile function of pure (3α,8α,13α)-E-41/HE-48 into the primary f-amide group of E-42/HE-49, besides partial epimerization at C-13;[9]: 100-103 [55]: 21,134-136 [3]: 150-151,169-170 an alternative procedure for the selective f-nitrile→f-amide conversion (BF3 in CH3COOH) later developed at Harvard proceeded without epimerization at C-13.[18]: 1569 footnote 62 [5]: 0:46:40-0:49:45 [55]: 21 A crystalline sample of the 3α,8α,13α-epimer of dicyano-cobalt (III)-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide E-42/HE-49, isolated by HPLC, was the first totally synthetic intermediate to be chromatographically and spectroscopically identified with a relay sample made from vitamin B12.[55]: 136-141 [3]: 170 In the remaining steps of the synthesis, only epimerization at C-13 played an important role,[55]: 19-21 with 13α being the configuration of the natural corrinoids, and 13β known as neo-epimers of vitamin B12 and its derivatives;[3]: 169-170 [81] these are readily separable by HPLC.[5]: 0:19:30-0:20:21 [55]: 135,208-209 In the course of 1972, comprehensive identifications (HPLC, UV/VIS, IR, NMR, CD, mass spectra) of crystalline samples of totally synthetic intermediates with the corresponding compounds derived from vitamin B12 were carried out in both laboratories: individually compared and identified were the 3α,8α,13α and 3α,8α,13β neo-epimer of f-amide E-42/HE-49, as well as the corresponding pair of C-13-epimeric nitriles E-41/HE-48.[55]: 206-221 [57]: 46-47 [5]: 0:27:28-0:46:32 All these dicyano-cobalt(III)-complexes are soluble in organic solvents[56]: 11 in which the separation power of HPLC by far exceeds that of analytical methods operating in water,[55]: 44-45 the solvent in which cobyric acid was to be identified, and where it exists as two easily equilibrating aquo-cyano complexes, epimeric regarding the position of the two non-identical axial Co ligands.[63]: 196-197 [57]: 49-60 These thorough identifications of the totally synthetic with partially synthetic materials mark the accomplishment of the two syntheses. They also reciprocally provided structure proof for a specific constitutional isomer isolated from a mixture of isomeric mono-amides formed in the partial ammonolysis of the B12-derived cobester,[note 9] tentatively assigned to be the 3α,8α,13α-f-amide E-42/HE-49 (see fig. 30).[56]: 9-18,67-70 [55]: 226-239 [59]  Figure 30: ETH/Harvard joint final steps: hexamethylcobyrinate-f-amide (synthesis and identification) to cobyric acid |
| Synthetic cobyric acid |
|---|
|
The final task of reaching cobyric acid from f-amide E-42/HE-49 required the critical step of hydrolysing the singular amide function into a free carboxyl function without touching any of the six methoxycarbonyl groups around the molecule's periphery. Since exploratory attempts by the conventional method of amide hydrolysis via nitrosation led to detrimental side reactions at the chromophore, a novel way of "hydrolysing" the f-amide group without touching the six methylester groups was conceived and explored at ETH: treatment of f-amide E-42/HE-49 (B12-derived relay material) with the unusual reagent α-chloro-propyl-(N-cyclohexyl)-nitrone[82] and AgBF4 in CH2Cl2, then with HCl in H2O/dioxane, and finally with dimethylamine in isopropanol afforded the f-acid E-43/HE-50 in 57% yield.[63]: 24-25,159-172 [3]: 170-172 [5]: 0:53:17-0:58:30 Sustained experimentations at Harvard eventually showed the nitrosation method to be successful (N2O4, CCl4, NaOAc) and to produce the f-carboxyl group even more effectively.[3]: 172-173 [5]: 0:58:19-0:59:15 It was also at Harvard that conditions for the last step were explored, conversion of all remaining ester groups into primary amide groups by ammonolysis. Liquid ammonia in ethylene glycol, in the presence of NH4Cl and the absence of oxygen, converted f-carboxy-hexamethylester E-43/HE-50 into f-carboxy-hexa-amide E-44/HE-51 (= cobyric acid).[3]: 173-175 [55]: 24 This was crystallised and shown both as the α-cyano-β-aquo and the α-aquo-β-cyano form to be chromatographically and spectroscopically identical with the corresponding forms of natural cobyric acid.[5]: 0:59:53-1:09:58 [3]: 175-176 [63]: 26-27,196-221 At Harvard, the transformation E-43/HE-50 → E-44/HE-51 was eventually carried out starting with f-amide that had been obtained by total synthesis via the A/B approach.[57]: 47-61 The ETH group contented itself with a corresponding f-amide → cobyric acid conversion and subsequent cobyric acid identification where the actual starting material f-amide was derived from vitamin B12.[55]: 22 [63]: 15 [12]: footnote 45 [18]: 1570-1571 |
Notes
- For a review about syntheses of corrins, see[27]; this includes more recent synthetic approaches to vitamin B12 by the groups of Stevens,[27]: 293-298 Jacobi,[27]: 298-300 and Mulzer,[27]: 300-301 as well as references to approaches by Todd or Cornforth (see also[45]: 261-268 ) preceding the efforts by Eschenmoser and Woodward.[18]: 1493-1496
- Formulae in figs. 4 and 6 illustrate the atom, ring, and side chain enumeration in corrins: "Nomenclature of Corrinoids". Pure and Applied Chemistry. 48 (4): 495–502. 1976. doi:10.1351/pac197648040495.
- The year 1964 refers to the first corrin synthesis of a pentamethylcorrin via A/B-cyclization by iminoester/enamine-C,C-condensation;[28] the heptamethylcorrin shown here (M = Co(CN)2) was prepared by the same ring closure method in 1967.[29]
- Friedrich, W.; Gross, G.; Bernhauer, K.; Zeller, P. (1960). "Synthesen auf dem Vitamin-B12-Gebiet. 4. Mitteilung. Partialsynthese von Vitamin B12". Helvetica Chimica Acta. 43 (3): 704–712. doi:10.1002/hlca.19600430314. For recent partial syntheses of vitamin B12 and coenzyme B12 from cobyric acid, see Widner, Florian J.; Gstrein, Fabian; Kräutler, Bernhard (2017). "Partial Synthesis of Coenzyme B12 from Cobyric Acid". Helvetica Chimica Acta. 100 (9): e1700170. doi:10.1002/hlca.201700170.
- See Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor in (Show/Hide) "Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach)".
- Letter from J. W. Cornforth to A. Eschenmoser, April 16th, 1984, see [18]: 1561 footnote 51 ; see also refs.[6][44]: 40 [45]: 265 . This preparation of a ring-C precursor from (+)-camphor involved 8 steps, compared to 4 steps[note 5] from the ETH ring-B precursor (but it used a commonly available precursor instead of "precious" material!)
- See Synthesis of the A-D-component carrying the propionic acid function at ring D as methoxycarbonyl group (model A-D-component) in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach".
- See Synthesis of the A-D-component carrying the propionic acid function at ring D as nitrile group in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach".
- Cobester (dicyano-Co-cobyrinic acid heptamethylester) is a non-natural cobyric acid derivative that had played an important subsidiary role in the B12 total syntheses;[55]: 14,21,51–90,222–260 it is prepared in one step from vitamin B12 by acid-catalyzed methanolysis.[56]: 9–18
- "University of Bristol. WILSON BAKER SYMPOSIUM: Previous Wilson Baker lectures" (PDF). Retrieved 2019-10-29. See also Eschenmoser lecture announcements in "Notizen". Nachrichten aus Chemie und Technik. 20 (5): 89–90. 1972. doi:10.1002/nadc.19720200502.
- Research reports of the Harvard postdoctoral fellows involved in the vitamin B12 synthesis are in the Harvard archives; see "Collection: Papers of Robert Burns Woodward, 1873-1980, 1930-1979 | HOLLIS for Archival Discovery". Retrieved 2019-10-29.
- The only "joint publication" is a 1972 interview with Eschenmoser and Woodward in Basle; [31] see also[18]: 1572–1574 [64]: 1478 .
- References given here are a selection from more than 60 publications where these epochal syntheses are discussed in more or less detail. They are also used to teach natural product synthesis in advanced courses or research group seminars, e.g., Eschenmoser, A. (2001). "Epilogue: Synthesis of Coenzyme B12: A Vehicle for the Teaching of Organic Synthesis". In Quinkert, Gerhard; Kisakürek, M. Volkan (eds.). Essays in Contemporary Chemistry: From Molecular Structure Towards Biology. Zürich: Verlag Helvetica Chimica Acta. pp. 391–441. doi:10.1002/9783906390451.ch12. ISBN 9783906390284. free version: Eschenmoser, Albert (2015). "Synthesen von Vitamin B12 (an die Hörer verteilte Unterlagen)". doi:10.3929/ethz-a-010521002. Retrieved 2021-01-06.
{{cite journal}}: Cite journal requires|journal=(help) - This is the only part of the Harvard contributions published with full experimental details so far: Fleming, Ian; Woodward, R. B. (1973). "A synthesis of (−)-(R)-trans-β-(1,2,3-trimethylcyclopent-2-enyl)acrylic acid". Journal of the Chemical Society, Perkin Transactions 1: 1653–1657. doi:10.1039/P19730001653. Fleming, Ian; Woodward, R. B. (1968). "Exo-2-Hydroxyepicamphor". Journal of the Chemical Society C: Organic: 1289–1291. doi:10.1039/J39680001289.
- This name of a left-hand side ("western half") building block relates to the Hesperides, the Nymphs of the West, as do Hesperidium and (the chemically completely unrelated) Hesperidin;[1] cf. other colorful namings by Woodward: pentacyclenone,[1]: 530 corrnorsterone;[1]: 534 corrigenolide, corrigenate: corrin-generating seco-corrins.[2]: 285,296 The ETH group had named its right-hand side building block "(thio)dextrolin" based on "dexter", Latin for "right".[1]: 538-539
- Camphorquinone is produced from camphor by reaction with selenium dioxide: see White, James D.; Wardrop, Duncan J.; Sundermann, Kurt F. (2002). Checked by Kenji Koga, Kei Manabe, Christopher E. Neipp, and Stephen F. Martin. "Camphorquinone and Camphorquinone Monoxime". Organic Syntheses. 79: 125. doi:10.15227/orgsyn.079.0125.
- Wick, Alexander: Report Part I, Harvard University 1967 (unpublished[note 11]), quoted in[44]: 38–39 .
- See Syntheses of the ring-B precursor in (Show/Hide) "Synthesis of the ETH B-C-component".
- See A/B-ring closure in (Show/Hide) "Coupling of Harvard A-D-components with the ETH B-C-component".
- See Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile (the common corrinoid intermediate) from the ring-D-differentiated A-D-component in (Show/Hide) "Coupling of Harvard A-D-components with the ETH B-C-component".
References
- Woodward, R. B. (1968). "Recent advances in the chemistry of natural products". Pure and Applied Chemistry. 17 (3–4): 519–547. doi:10.1351/pac196817030519. PMID 5729287.
- Woodward, R. B. (1971). "Recent advances in the chemistry of natural products". Pure and Applied Chemistry. 25 (1): 283–304. doi:10.1351/pac197125010283. PMID 5095424.
- Woodward, R. B. (1973). "The total synthesis of vitamin B12". Pure and Applied Chemistry. 33 (1): 145–178. doi:10.1351/pac197333010145. PMID 4684454.
- Woodward, Robert B. (November 27, 1972). R.B. Woodward Total Synthesis of Vitamin B12 Lecture - Part 1 (recorded lecture). Introduction by David Dolphin. Harvard University, Cambridge MA (U.S.A.): YouTube. Archived from the original on 2021-12-21. Retrieved 2020-01-25.
- Woodward, Robert B. (November 27, 1972). R.B. Woodward Total Synthesis of Vitamin B12 Lecture - Part 2 (recorded lecture). Harvard University, Cambridge MA (U.S.A.): YouTube. Archived from the original on 2021-12-21. Retrieved 2020-01-25.
- Eschenmoser, A. (1968). "Die Synthese von Corrinen". Moderni Sviluppi della Sintesi Organica (X Corso estivo di chimica, Fondazione Donegani, Frascati 25.9.-5.10.1967) (in German). Roma: Accademia Nazionale dei Lincei. pp. 181–214. ISBN 8821804054. ISSN 0515-2216.
- Eschenmoser, A. (1968). "Current Aspects of Corrinoid Synthesis". Proceedings of the Robert A. Welch Foundation Conference on Chemical Research. 12: 9–47. doi:10.3929/ethz-b-000467558. ISSN 0557-1588.
- Eschenmoser, A. (1970). "Centenary Lecture (Delivered November 1969). Roads to corrins". Quarterly Reviews, Chemical Society. 24 (3): 366–415. doi:10.1039/qr9702400366.
- Eschenmoser, A. (1971). Studies on Organic Synthesis. XXIIIrd International Congress of Pure and Applied Chemistry: special lectures presented at Boston, USA, 26-30 July 1971. Vol. 2. London: Butterworths. pp. 69–106. doi:10.3929/ethz-a-010165162. hdl:20.500.11850/84699. ISBN 0-408-70316-4.
- Fuhrer, W.; Schneider, P.; Schilling, W.; Wild, H.; Schreiber, J.; Eschenmoser, A. (1972). "Totalsynthese von Vitamin B12: die photochemische Secocorrin-Corrin-Cycloisomerisierung". CHIMIA (abstract of lecture). 26: 320.Maag, H.; Obata, N.; Holmes, A.; Schneider, P.; Schilling, W.; Schreiber, J.; Eschenmoser, A. (1972). "Totalsynthese von Vitamin B12: Endstufen". CHIMIA (abstract of lecture). 26: 320.
- Eschenmoser, A. (1974). "Organische Naturstoffsynthese heute. Vitamin B12 als Beispiel". Die Naturwissenschaften. 61 (12): 513–525. Bibcode:1974NW.....61..513E. doi:10.1007/BF00606511. PMID 4453344. S2CID 45688091.
- Eschenmoser, A.; Wintner, C. (1977). "Natural product synthesis and vitamin B12". Science. 196 (4297): 1410–1420. Bibcode:1977Sci...196.1410E. doi:10.1126/science.867037. PMID 867037.
- Zass, E. (2014). "Of a Landmark Total Synthesis yet Unpublished in Full Experimental Detail - Vitamin B12 (Slides of the Skolnik Award Lecture at the 248th ACS National Meeting, San Francisco CA, Aug. 12, 2014)". SlideShare. LinkedIn. Retrieved 2020-01-25. See also Warr, Wendy (2014). "Herman Skolnik Award Symposium Honoring Engelbert Zass". Chemical Information Bulletin. 66 (4/Winter 2014): 37–40. Retrieved 2020-01-25.
- Craig, G. Wayne (2016). "Total synthesis of vitamin B12 - a fellowship of the ring". Journal of Porphyrins and Phthalocyanines. 20: 1–20. doi:10.1142/S1088424615500960.
- Nicolaou, K. C.; Sorensen, E. J. (1996). "Chapter 8: Vitamin B12. R. B. Woodward and A. Eschenmoser (1973)". Classics in Total Synthesis: Targets, Strategies, Methods. Weinheim: VCH Verlag Chemie. pp. 99-136. ISBN 978-3-527-29231-8.
- Marko, I. E. (2001). "Natural product synthesis: the art of total synthesis". Science. 294 (5548): 1842–1843. doi:10.1126/science.1067545. PMID 11729290. S2CID 22467000.
- Eschenmoser, A. (2001). "RBW, Vitamin B12, and the Harvard-ETH Collaboration". In Benfey, O. Theodor; Morris, Peter J. T. (eds.). Robert Burns Woodward - Architect and Artist in the World of Molecules. History of Modern Chemical Sciences series. Philadelphia: Chemical Heritage Foundation. pp. 23–38. ISBN 978-0941901253. ISSN 1069-2452.
- Eschenmoser, Albert (2015). "Corrin Syntheses. Part I". Helvetica Chimica Acta. 98 (11–12): 1483–1600. doi:10.1002/hlca.201400277.
- Nicolaou, K. C.; Sorensen, E. J.; Winssinger, N. (1998). "The Art and Science of Organic and Natural Products Synthesis". Journal of Chemical Education. 75 (10): 1225–1258. Bibcode:1998JChEd..75.1225N. doi:10.1021/ed075p1225.
- Nicolaou, K. C.; Vourloumis, Dionisios; Winssinger, Nicolas; Baran, Phil S. (2000). "The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century". Angewandte Chemie International Edition. 39 (1): 44–122. doi:10.1002/(SICI)1521-3773(20000103)39:1<44::AID-ANIE44>3.0.CO;2-L. PMID 10649349.
- Eschenmoser, Albert (1988). "Vitamin B12: Experiments Concerning the Origin of Its Molecular Structure". Angewandte Chemie International Edition in English. 27: 5–39. doi:10.1002/anie.198800051.
- Brink-Shoemaker, Clara; Cruickshank, D. W. J.; Crowfoot Hodgkin, Dorothy; Kamper, M. Jennifer; Pilling, Diana (1964). "The structure of Vitamin B12 VI. The structure of crystals of vitamin B12 grown from and immersed in water". Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences. 278 (1372): 1–26. Bibcode:1964RSPSA.278....1B. doi:10.1098/rspa.1964.0042. S2CID 93447375.
- "CSD Entry: VITAMB Vitamin B12 hydrate". Cambridge Structural Database CCDC/Fiz Karlsruhe. Retrieved 2020-12-24.
- Hodgkin, Dorothy Crowfoot; Kamper, Jennifer; MacKay, Maureen; Pickworth, Jenny; Trueblood, Kenneth N.; White, John G. (1956). "Structure of Vitamin B12". Nature. 178 (4524): 64–66. Bibcode:1956Natur.178...64H. doi:10.1038/178064a0. PMID 13348621. S2CID 4210164.
- Glusker, Jenny P. (1995). "Vitamin B12 and the B12 Coenzymes". Vitamins and Hormones. 50: 1–76. doi:10.1016/S0083-6729(08)60654-8. ISBN 9780127098500. PMID 7709599.
- Bernhauer, K.; Dellweg, H.; Friedrich, W.; Gross, Gisela; Wagner, F. (1960). "Notizen: Vitamin B12-Faktor V1a, ein neuer "inkompletter" Grundkörper der Vitamin B12-Gruppe". Zeitschrift für Naturforschung B. 15 (5): 336–337. doi:10.1515/znb-1960-0522. S2CID 98606543.
- Montforts, Franz-Peter; Osmers, Martina; Leupold, Dennis (2012). "Chemical Synthesis of Artificial Corrins". In Kadish, Karl M.; Smith, Kevin M.; Guilard, Roger (eds.). Handbook of Porphyrin Science. Vol. 25. World Scientific Publishing. pp. 265–307. doi:10.1142/9789814397605_0020. ISBN 978-981-4397-66-7.
- Bertele, E.; Boos, H.; Dunitz, J. D.; Elsinger, F.; Eschenmoser, A.; Felner, I.; Gribi, H. P.; Gschwend, H.; Meyer, E. F.; Pesaro, M.; Scheffold, R. (1964). "A Synthetic Route to the Corrin System". Angewandte Chemie International Edition in English. 3 (7): 490–496. doi:10.1002/anie.196404901.
- Felner-Caboga, I.; Fischli, A.; Wick, A.; Pesaro, M.; Bormann, D.; Winnacker, E.L.; Eschenmoser, A. (1967). "rac.-Dicyano-(1,2,2,7,7,12,12-heptamethylcorrin)-cobalt(III)". Angewandte Chemie International Edition in English. 6 (10): 864–866. doi:10.1002/anie.196708643.
- Benfey, O. Theodor; Morris, Peter J. T., eds. (2001). Robert Burns Woodward - Architect and Artist in the World of Molecules. History of Modern Chemical Sciences series. Philadelphia: Chemical Heritage Foundation. ISBN 978-0941901253. ISSN 1069-2452.
- ""Herr Woodward bedauert, daß die Sache fertig ist." Woodward und Eschenmoser über Vitamin B12 und die Situation der organischen Chemie". Nachrichten aus Chemie und Technik. 20 (8): 147–150. 2010. doi:10.1002/nadc.19720200804.
- Krieger, J. H. (1973). "Vitamin B12: the struggle toward synthesis". Chemical & Engineering News. 51 (11/March 12): 16–29. doi:10.1021/cen-v051n011.p016.
- Craig, G. Wayne (2014). "Eschenmoser approach to vitamin B12 by A/D strategy". Resonance. 19 (7): 624–640. doi:10.1007/s12045-014-0064-4. S2CID 118161709.
- Smith, K. M. (1971). "Recent developments in the chemistry of pyrrolic compounds". Quarterly Reviews, Chemical Society. 25: 31–85. doi:10.1039/qr9712500031.
- Bertele, Erhard; Scheffold, Rolf; Gschwend, Heinz; Pesaro, Mario; Fischli, Albert; Roth, Martin; Schossig, Jürgen; Eschenmoser, Albert (2015). "Corrin Syntheses. Part IV". Helvetica Chimica Acta. 98 (11–12): 1755–1844. doi:10.1002/hlca.201200342.
- Yamada, Yasuji; Miljkovic, D.; Wehrli, P.; Golding, B.; Löliger, P.; Keese, R.; Müller, K.; Eschenmoser, A. (1969). "A New Type of Corrin Synthesis". Angewandte Chemie International Edition in English. 8 (5): 343–348. doi:10.1002/anie.196903431. PMID 4977933.
- Yamada, Yasuji; Wehrli, Pius; Miljkovic, Dusan; Wild, Hans-Jakob; Bühler, Niklaus; Götschi, Erwin; Golding, Bernard; Löliger, Peter; Gleason, John; Pace, Brian; Ellis, Larry; Hunkeler, Walter; Schneider, Peter; Fuhrer, Walter; Nordmann, René; Srinivasachar, Kasturi; Keese, Reinhart; Müller, Klaus; Neier, Reinhard; Eschenmoser, Albert (2015). "Corrin Syntheses. Part VI". Helvetica Chimica Acta. 98 (11–12): 1921–2054. doi:10.1002/hlca.201500012.
- Stevens, R. V. (1982). "The Total Synthesis of Vitamin B12". In Dolphin, D. (ed.). Vitamin B12. Vol. 1. New York: John Wiley & Sons. pp. 169–200. ISBN 978-0-471-03655-5.
- Wild, Jost (1964). Synthetische Versuche in Richtung auf natürlich vorkommende Corrinoide (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3492). doi:10.3929/ethz-a-000088927. hdl:20.500.11850/132003.
- Woodward, R. B. (1963). "Versuche zur Synthese des Vitamins B12". Angewandte Chemie. 75 (18): 871–872. Bibcode:1963AngCh..75..871W. doi:10.1002/ange.19630751827.
- Woodward, R. B. (1967). "The conservation of orbital symmetry". Aromaticity. Chemical Society Special Publication. Vol. 21. London: Royal Society of Chemistry. pp. 217–249.
- Money, T. (1985). "Camphor: A chiral starting material in natural product synthesis". Natural Product Reports. 2 (3): 253–289. doi:10.1039/np9850200253. PMID 3906448.
- Locher, Urs (1964). Darstellung eines Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3611). doi:10.3929/ethz-a-000090323. hdl:20.500.11850/131398.
- Dubs, Paul (1969). Beiträge zur Synthese von Vitamin B12: Darstellung vinyloger Amidine mit der Sulfidkontraktions-Methode (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4297). doi:10.3929/ethz-a-000093384. hdl:20.500.11850/133822.
- Jackson, A. H.; Smith, K. M. (1973). "The Total Synthesis of Pyrrole Pigments". In Apsimon, John (ed.). Total Synthesis of Natural Products. Vol. 1. pp. 143–278. doi:10.1002/9780470129647.ch3. ISBN 9780471032519.
- Löliger, Peter (1968). Darstellung eines die Ringe B und C umfassenden Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4074). doi:10.3929/ethz-a-000093406. hdl:20.500.11850/133844.
- Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. (1971). "Sulfidkontraktion via alkylative Kupplung: Eine Methode zur Darstellung von β-Dicarbonylderivaten. Über synthetische Methoden, 1. Mitteilung". Helvetica Chimica Acta. 54 (2): 710–734. doi:10.1002/hlca.19710540229.
- Schneider, Peter (1972). Totalsynthese von Derivaten des Dicyano-cobalt(III)-5,15-bis-nor-cobyrinsäure-hepta-methylesters (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4819). doi:10.3929/ethz-a-000090603. hdl:20.500.11850/132893.
- Eschenmoser, A. (1994). "B12: reminiscences and afterthoughts". In Chadwick, Derek J.; Ackrill, Kate (eds.). The Biosynthesis of the Tetrapyrrole Pigments. Ciba Foundation Symposium 180 (Novartis Foundation Symposia 105). Chichester: J. Wiley & Sons. pp. 309–336. ISBN 978-0471939474.
- Eschenmoser, A. (1969). "The role of transition metals in the chemical synthesis of corrins". Pure and Applied Chemistry. 20 (1): 1–23. doi:10.1351/pac196920010001.
- Fuhrer, Walter (1973). Totalsynthese von Vitamin B12: Der photochemische Weg (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5158). doi:10.3929/ethz-a-000086601. hdl:20.500.11850/131362.
- Huber, J. F. K. (1969). "High Efficiency, High Speed Liquid Chromatography in Columns". Journal of Chromatographic Science. 7 (2): 85–90. doi:10.1093/chromsci/7.2.85.
- Schreiber, J. (1971). "Ein Beispiel zur Anwendung der schnellen Flüssigchromatogarphie in der organischen Synthese". CHIMIA. 25 (12): 405–407.
- Hertzog, D. (1973). "Utilisation de la chromatographie liquide haute pression en synthese organique". Informations Chimique. 119: 229–231.
- Maag, Hans (1973). Totalsynthese von Vitamin B12: Dicyano-Co(III)-Cobyrinsäure-Hexamethylester-f-Amid (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5173). doi:10.3929/ethz-a-000085446. hdl:20.500.11850/131110.
- Werthemann, Lucius (1968). Untersuchungen an Kobalt(II)- und Kobalt(III)-Komplexen des Cobyrinsäure-heptamethylesters (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4097). doi:10.3929/ethz-a-000093488. hdl:20.500.11850/133926.
- Woodward, R. B. (1979). "Synthetic Vitamin B12". In Zagalak, B.; Friedrich, W. (eds.). Vitamin B12 (Proceedings of the 3rd European Symposium on Vitamin B12 and Intrinsic Factor, University of Zurich, March 5-8, 1979). Berlin: W. de Gruyter. pp. 37–87. doi:10.1515/9783111510828-005. ISBN 3-11-007668-3.
- Wintner, Claude E. (2006). "Recollecting the Institute of Organic Chemistry, ETH Zürich, 1972-1990". CHIMIA. 60 (3): 142–148. doi:10.2533/000942906777675029.
- Ernst, Ludger; Maag, Hans (2006). "Preparation and Structure Proof of the Four Isomeric Dicyanocobyrinic Acid Hexamethyl Ester Monoamides Carrying the Amide Group on a Propionic Acid Side Chain". Liebigs Annalen. 1996 (3): 323–326. doi:10.1002/jlac.199619960306.
- Wick, Alexander (1964). Untersuchungen in Richtung einer Totalsynthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3617). doi:10.3929/ethz-a-000090041. hdl:20.500.11850/132537.
- Wiederkehr, René (1968). Darstellung von Zwischenprodukten zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4239). doi:10.3929/ethz-a-000087656. hdl:20.500.11850/131502.
- Huber, Willy (1969). Beiträge zur Synthese von Vitamin B12: Zum Problem der (C-D)-Verknüpfung (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4298). doi:10.3929/ethz-a-000090323. hdl:20.500.11850/132700.
- Schilling, Walter (1974). Totalsynthese von Vitamin B12. Darstellung von Zwischenprodukten und partialsynthetische Endstufen (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5352). doi:10.3929/ethz-a-000085344. hdl:20.500.11850/131064.
- Eschenmoser, Albert (2015). "Introductory Remarks on the Publication Series 'Corrin Syntheses-Parts I-VI'". Helvetica Chimica Acta. 98 (11–12): 1475–1482. doi:10.1002/hlca.201400399.
- Scheffold, Rolf; Bertele, Erhard; Gschwend, Heinz; Häusermann, Werner; Wehrli, Pius; Huber, Willi; Eschenmoser, Albert (2015). "Corrin Syntheses. Part II". Helvetica Chimica Acta. 98 (11–12): 1601–1682. doi:10.1002/hlca.201200095.
- Pesaro, Mario; Elsinger, Fritz; Boos, Helmut; Felner-Caboga, Ivo; Gribi, Hanspeter; Wick, Alexander; Gschwend, Heinz; Eschenmoser, Albert (2015). "Corrin Syntheses. Part III". Helvetica Chimica Acta. 98 (11–12): 1683–1754. doi:10.1002/hlca.201200308. Blaser, Hans-Ulrich; Winnacker, Ernst-Ludwig; Fischli, Albert; Hardegger, Bruno; Bormann, Dieter; Hashimoto, Naoto; Schossig, Jürgen; Keese, Reinhart; Eschenmoser, Albert (2015). "Corrin Syntheses. Part V". Helvetica Chimica Acta. 98 (11–12): 1845–1920. doi:10.1002/hlca.201300064.
- "ETH Research Collection (previously ETH e-collection)". ETH Zurich. Retrieved 2020-01-25.
- Goto, Toshio (1975). "chapter 11.34: Synthesis of vitamin B12". In Nakanishi, Koji; Goto, Toshio; Sho, Ito; Natori, Shinsaku; Nozoe, Shigeo (eds.). Natural Products Chemistry. Vol. 2. Tokyo: Kodansha/Academic Press. pp. 480–496. ISBN 0-12-513902-0.
- Riether, Doris; Mulzer, Johann (2003). "Total Synthesis of Cobyric Acid: Historical Development and Recent Synthetic Innovations". European Journal of Organic Chemistry. 2003: 30–45. doi:10.1002/1099-0690(200301)2003:1<30::AID-EJOC30>3.0.CO;2-I.
- Corey, E. J.; Chow, S. W.; Scherrer, R. A. (1957). "The synthesis of α-santalene and of trans-Δ11,12-iso-α-santalene". Journal of the American Chemical Society. 79 (21): 5773–5777. doi:10.1021/ja01578a049. Guha, P. C.; Bhattachargya, S. C. (1944). "Santalol series. II. Synthesis of d- and dl-π-hydroxycamphor, d- and dl-teresantalol, and d- and dl-tricycloekasantalic acid". Journal of the Indian Chemical Society. 21: 271–280. Corey, E. J.; Ohno, Masaji; Chow, S. W.; Scherrer, Robert A. (1959). "Acid-catalyzed cleavage of π-substituted tricyclenes. Synthesis of 3,8-cyclocamphor". Journal of the American Chemical Society. 81 (23): 6305–6309. doi:10.1021/ja01532a048. Hasselstrom, Torsten (1931). "Studies on π-camphor derivatives. II. The identity of dihydro-teresantalic acid with 7-π-apocamphan-carboxylic acid". Journal of the American Chemical Society. 53 (3): 1097–1103. doi:10.1021/ja01354a043.
- Kaski, B. A. (1971). Studies on Packing in Molecular Crystals (PhD). Harvard University. pp. II-1.
- Blaser, Hans-Ulrich (1971). Herstellung und Eigenschaften eines metallfreien Corrin-Derivates (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4662). doi:10.3929/ethz-a-000091385. hdl:20.500.11850/133210.
- Fischli, Albert (1968). Die Synthese metallfreier Corrine (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4077). doi:10.3929/ethz-a-000267791. hdl:20.500.11850/137445.
- Jauernig, D.; Rapp, P.; Ruoff, G. (1973). "5-Nor-, 15-Nor- und 5,15-Dinorcorrinoide". Hoppe-Seyler's Zeitschrift für physiologische Chemie. 354 (8): 957–966. doi:10.1515/bchm2.1973.354.2.957.
- Manasse, O.; Samuel, E. (1902). "Reactionen des Campherchinons". Berichte der Deutschen Chemischen Gesellschaft. 35 (3): 3829–3843. doi:10.1002/cber.190203503216.
- Wick, A. E.; Felix, Dorothee; Steen, Katharina; Eschenmoser, A. (1964). "Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit Hilfe von Acetalen des N,N-Dimethylacetamids. Vorläufige Mitteilung". Helvetica Chimica Acta. 47 (8): 2425–2429. doi:10.1002/hlca.19640470835. Felix, Dorothee; Gschwend-Steen, Katharina; Wick, A. E.; Eschenmoser, A. (1969). "Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit 1-Dimethylamino-1-methoxy-äthen". Helvetica Chimica Acta. 52 (4): 1030–1042. doi:10.1002/hlca.19690520418.
- Johnson, William Summer; Werthemann, Lucius; Bartlett, William R.; Brocksom, Timothy J.; Li, Tsung-Tee; Faulkner, D. John; Petersen, Michael R. (1970). "Simple stereoselective version of the Claisen rearrangement leading to trans-trisubstituted olefinic bonds. Synthesis of squalene". Journal of the American Chemical Society. 92 (3): 741–743. doi:10.1021/ja00706a074. Ireland, Robert E.; Mueller, Richard H.; Willard, Alvin K. (1976). "The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation". Journal of the American Chemical Society. 98 (10): 2868–2877. doi:10.1021/ja00426a033.
- Wild, Hans-Jakob (1972). Die Synthese von Corrin-Komplexen durch photochemische A/D-Cycloisomerisierung (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4848). doi:10.3929/ethz-a-000090212. hdl:20.500.11850/132648.
- Gardiner, Maureen; Thomson, Andrew J. (1974). "Luminescence properties of some synthetic metallocorrins". Journal of the Chemical Society, Dalton Transactions (8): 820–828. doi:10.1039/DT9740000820.
- Winnacker, Ernst-Ludwig (1968). Ligandreaktivität synthetischer Kobalt(III)-Corrin-Komplexe (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4177). doi:10.3929/ethz-a-000150375. hdl:20.500.11850/136417.
- Bonnett, R.; Godfrey, J. M.; Math, V. B. (1971). "Cyano-13-epicobalamin (Neovitamin B12) and its relatives". Journal of the Chemical Society C: Organic. 22: 3736–43. doi:10.1039/j39710003736. PMID 5167083.
- Kempe, U. M.; Das Gupta, T. K.; Blatt, K.; Gygax, P.; Felix, Dorothee; Eschenmoser, A. (1972). "α-Chlor-nitrone I: Darstellung und Ag+-induzierte Reaktion mit Olefinen. Über synthetische Methoden, 5. (Vorläufige) Mitteilung". Helvetica Chimica Acta. 55 (6): 2187–2198. doi:10.1002/hlca.19720550640.